Journal of Alzheimers & Neurodegenerative Diseases Category: Clinical
Type: Review Article
Fundamental Neurovascular Components for the Development of Complex and Dynamic in Vitro Brain Equivalent Models
*Corresponding Author(s):
Goodwell NzouWake Forest Institute For Regenerative Medicine, Wake Forest School Of Medicine, Winston-Salem. NC 27101, United States
Tel:+15852107532,
Email:gnzou@wakehealth.edu
Received Date: May 06, 2019
Accepted Date: Jun 17, 2019
Published Date: Jun 24, 2019
Abstract
The lack of in vitro human brain equivalent models with functional neurons and other supporting cell types of the human cortex impedes scientific understanding of neurologic disease progression and has significantly limited drug development. These cell types (astrocytes, microglia, oligodendrocytes, neurons, pericytes, and microvascular endothelial cells) interact in concert to form a tissue unit termed the neurovascular unit (NVU). Critical to the normal function of the NVU is the formation and maturation of a Blood Brain Barrier (BBB) which is one of the primary barriers for CNS targeting investigational drugs. Understanding these cellular interactions is essential to the development of drugs to treat a wide array of diseases and neurological disorders. Current two- and three-dimensional in vitro models, in addition to in vivo animal models, often fail to mimic the physiological properties of the human NVU because most contain cells of varying species that do not accurately model human brain physiology. In addition, they contain synthetic components not representative of the environment within the human cortex. We hereby describe the fundamental components of the neurovascular unit necessary for developing complex and dynamic in vitro models that could be implemented in pre-clinical studies and disease modeling. We emphasize the importance of human-derived brain cells for translational relevancy. We hope that the inclusion of these components will overcome some of the limitations of current 2D in vitro models and may have applications in drug discovery and neurotoxicity testing.
Keywords
Alzheimer’s disease; Brain; Microglia
SIGNIFICANCE
Our lab developed a neurovascular unit model that is comprised of only human cells, which better represents human brain physiology in a more rapid and scalable design than current in vitro and in vivo models. The incorporation of essential components of the neurovascular unit - endothelial cells, pericytes, astrocytes, microglia, oligodendrocytes and neurons - mean that the model can be used not only to study the BBB mechanisms and functions, but also to evaluate the effect of drugs that cross the BBB on other human brain cell types within the NVU. This work can, therefore, assist in the validation of central nervous system safety of candidate molecules designed for both neurological and non-neurological diseases. The model may also provide a platform for evaluating neurodegenerative disease biomarkers and possibly disease pathologies with proper organoid maturation.
BACKGROUND
In the Unites States alone, 45 million people are affected by one of the 12 most prevalent neurologic disorders, while over one million adults are newly diagnosed with brain diseases or disorders [1]. Yet, there are limited treatment options, resulting in significant economic and social costs. For example, the Alzheimer’s Association reported that in 2016 alone, the national cost for Alzheimer’s and other dementias was approximately $236 billion [2]. The discovery of effective therapies has been limited by the low success rate of investigational drug trials, which in part is due to the lack of human brain-equivalent models [3]. The development of in vitro models that closely mimic human brain tissue remains a challenge. Current two- and three-dimensional tissue culture methods that have been described in drug screening and disease models, in addition to current animal models, do not mimic human physiology because they either consist of animal cells or do not contain all the necessary components that are critical to the normal function of the human neurovascular unit (NVU)[4-18]. Some models also utilize extracellular matrix (ECM) components or artificial, though biocompatible, membranes that are not present in the adult brain. This makes it more challenging to translate the results from these models to human disease applications. Here, we describe a model that incorporate six cell types found within the human brain cortex and we highlight their respective roles in maintaining normal function of the NVU by regulating blood-brain barrier (BBB) integrity. The six cell types discussed below are: human brain microvascular endothelial cells (HBMVEC), human pericytes (HBVP), human astrocytes (HA), human microglia (HM), human oligodendrocytes (HO), and human neurons (HN). We will also discuss the contribution of the extracellular matrix to the basement membrane and to the BBB integrity that is central to normal function of the NVU. We will briefly discuss BBB dysfunction in a few neurological disorders to highlight the value of incorporating the above-named components in understanding their impact in creating a dynamic disease model. We will conclude with a brief description and perspectives on the significance of the findings from a model developed in our lab.
Current in vivo and in vitro models
The use of animal models and in vitro models consisting of rodent cells fails to acknowledge the lack genetic correlation between humans and rodents [19]. Even though two-dimensional in vitro co-culture systems provide a platform for studying the interaction of cells, and the formation and some functions of the blood brain barrier [9], animal or human cells in two dimensional platforms still do not recapitulate the three-dimensional microenvironment within the human NVU.
Cell type limitations in current models
The number of different cell types that can be used in co-culture models while maintaining specific cell localization is limited in two dimensional systems. Critical to the maintenance of the blood brain barrier in NVU is the interaction between all six cell types in our model. Tight and adherens junction protein formation between adjacent human brain microvascular endothelial cells (HBMVEC) plays a central role in regulating transport of nutrients into the brain tissue. Brain derived neurotrophic factor secreted by HBMVECs support proliferation and survival of oligodendrocytes in the NVU [20]. The endothelial cells release platelet-derived growth factor (PDGF) to recruit pericytes, which in turn produce ECM necessary for the formation of the basement membrane. Additionally, it is well known that pericytes cover approximately 30% of the neuro-microvasculature and play a role in contractile function [21], microvasculature maturation [22] and collagen production [23]. The association of pericytes to blood vessels has also been suggested to regulate endothelial cell proliferation, migration, differentiation, survival, and vascular branching [24]. This illustrates their direct interaction with endothelial cells and their crucial role in maintaining the integrity of the BBB in the NVU [25,26]. Glial and neuronal cells induce the BBB by upregulating membrane associated enzymes [27-29]. Yet some models only consist of endothelial cells and neurons [30]. Often, models that incorporate neurons do not include oligodendrocytes that are crucial for neuronal network function and for myelination [31,32]. Those models ignore the importance of myelination in normal impulse conduction. Moreover, the interaction between astrocytes and oligodendrocytes is well documented [31], and astrocytes are known to influence the integrity of the BBB [33-35]. More importantly, oligodendrocytes are known to secret transforming growth factor beta (TGF-β), a factor known to promote BBB integrity through the activation of the MEK/ERK pathway, which consequently upregulates expression of tight junction proteins [36]. A significant body of evidence exists, both in vitro and in vivo, describing the astrocyte interaction with the cerebral endothelium and the mechanism by which this helps determine BBB function, morphology (i.e. tightness), and protein expression [28,37-39]. This dynamic interaction between cell types is crucial for the normal function of a NVU. A detailed look at the contribution of each of the cellular components and other components to the normal function of the NVU in the context of the blood brain barrier is outlined below.
Neurovascular unit
A brain tissue unit consisting of endothelial cells, pericytes, astrocytes, microglia, oligodendrocytes, neurons, and extracellular matrix components that make up the basement membrane is known as the neurovascular unit (NVU) [40]. Neurovascular coupling within the NVU represents the interaction between neurons and the vascular supply that provides them with necessary nutrients for proper neuronal function. The brain is dependent on constant blood supply. A healthy connection between neurons and the blood supply is critical for proper regulation of signals that control vascular changes. Neurovascular unit dysfunction that leads to an unmatched metabolic requirements in parenchymal tissue is evident in many pathological conditions including hypertension, Alzheimer’s disease, and ischemic stroke [41]. As shown in Figure 1, the position of astrocytes within the neurovascular unit is critical and unique for neurovascular coupling. Theoretical evidence suggests astrocytes contribute to functional hyperemia by shunting vasoactive stimuli from neuronal synapses to the astrocytic end feet that connects with the blood vessels [41]. Furthermore, the neuronal isoform of nitric oxide synthase produces a vasodilator that is linked to increase in cerebral blood flow [42-44]. These close interactions between neurons, glia and vascular cells defines and determines the normal functions of the NVU.
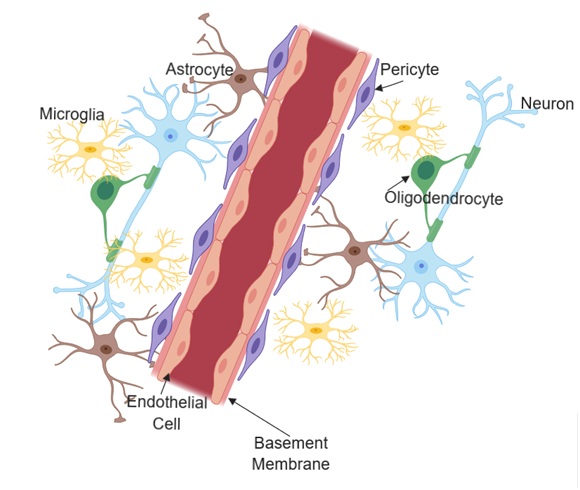 Figure 1: Structural and cellular composition of the neurovascular unit.
Figure 1: Structural and cellular composition of the neurovascular unit.The blood-brain barrier
Critical to the normal function of the neurovascular unit is the blood-brain barrier (BBB). It consists of a structurally organized endothelial cell lining that interacts with pericytes, astrocytes and the basement membrane to shield parenchymal brain tissue from toxins and pathogens while allowing specific nutrients to pass and providing a chemical composition for proper glial and neuronal function [45-51]. The delivery of essential nutrients such as oxygen and glucose, the removal of metabolic wastes, and the mediation of signaling of the endocrine glands is regulated at the BBB level [51-54]. (Figure 2)below depicts pathways by which different substances cross the BBB (steps 1-6). The figure also highlights the interaction between cells of the BBB with other brain cells. The specific functions of each of the components of the neurovascular unit with respect to the BBB are discussed below.
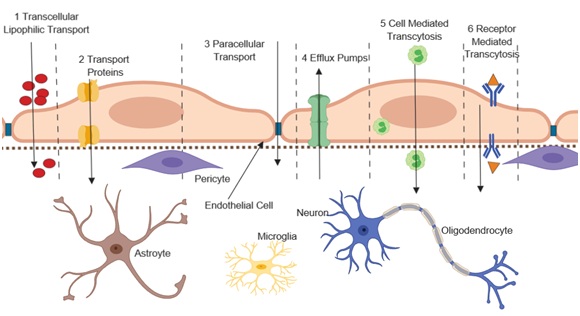 Figure 2: Transport pathways at the Blood Brain Barrier.
Figure 2: Transport pathways at the Blood Brain Barrier. FUNCTIONS OF NEUROVASCULAR UNIT COMPONENTS
Human brain microvascular endothelial cells
Brain endothelial cells have several peculiar properties that they do not share with peripheral endothelial cells. Brain endothelial cells receive and send signals to neighboring brain cells to enhance barrier properties as shown in Figure 3. Some of these properties include the lack of fenestrations, reduced transcytosis, and increased expression of specialized junctional proteins that limit paracellular transport between the luminal and the abluminal compartments. The junctional proteins include complexes of transmembrane proteins such as claudins, occludin and junctional adhesion molecules.
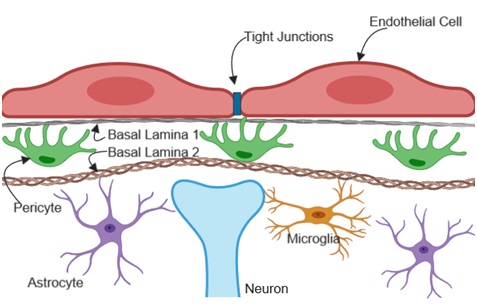
Figure 3: Brain endothelial cell interaction with parenchymal cells.
BBB properties exhibited by brain endothelial cells is mainly controlled by tight junctions containing junctional adhesion molecules arranged in a polarized manner between adjacent brain endothelial cells as shown if Figure 4. Junctional adhesion molecules are located on the luminal side while cadherins are on the abluminal side. Between the luminal and the abluminal are the tight junction proteins that creates a tight seal preventing free paracellular transport. Claudins are the main tight junctional protein that define BBB maturation [55-57]. Brain endothelial cells mainly express claudin 1, 3, 5 and 12 [57-60]. These tetraspan transmembrane proteins assist in the construction of the BBB and hence the maintenance of barrier integrity between adjacent endothelial cells [55]. Claudin 5 is regulated by β-catenin, however, it is inhibited by β-catenin when the transcriptional factor FOXO-1 that is induced by VEGF signaling is active in brain endothelial cells [56,61]. This regulation in endothelial cells determines BBB maturation.
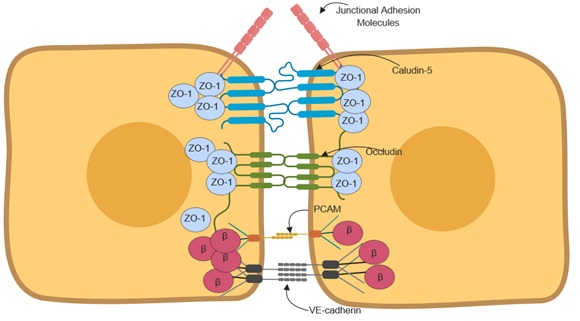
Figure 4: Junctional polarization and arrangement in adjacent endothelial cells.
Even though occludin expression in brain endothelial cells has been shown to decrease barrier integrity in vitro [62,63], occludin deficient mice had normal barrier function and the tight junction morphology was maintained [64].
The tight junctions (claudins and occludin) are anchored to the actin cytoskeleton by the Zonula occludens. Specifically, Zonula occluden-1 (ZO-1) is a membrane-associated granulate kinase like protein. ZO-1 stabilization of tight junctions is critical such that its deletion leads to increased permeability due to the disruption of the tight junction and redistribution of active myosin II [65].
Interestingly, barrier formation is evident between three adjoining endothelial cells as well. Specifically, tricellulin [66,67] and lipolysis stimulated lipoprotein receptor [68] are located at the point of connection between three cells. These are weak tri-cellular junctions; however, they stabilize the specialized junctions in epithelial cells.
Vascular endothelial cadherin (VE-cadherin) in endothelial cells controls permeability and also prevents leukocyte extravasation into the brain parenchyma [69]. Furthermore, VE- cadherin and N- cadherin function as adhesion receptors and are involved in downstream signaling via complexes of proteins bound to their cytosolic tails. N-cadherin in particular, mediates cell-cell interaction between endothelial cells and pericytes [70]. Β-catenin is also expressed by endothelial cells and is implicated in barrier functions. Its function as a co-transcriptional factor are critical in protein expression of claudin-5 [71], but its concerted anchoring of VE-cadherin with its homologue plakoglobin/γ-catenin to actin microfilaments stabilizes the junctional proteins and thereby improves barrier properties [71].
Brain endothelial cells have relatively large quantities and volumes of mitochondria compared to peripheral endothelial cells. This is because they contain enzymes and active transport systems that rely either directly on ATP consumption or on a secondary active transport systems that depend on the electrochemical gradient generated by active transporters [72]. Facilitated and active transporters at the BBB include glucose transporter-1 (GLUT-1) [73-75], permeability glycoprotein- (P-gp) also known as multidrug resistant protein 1 (Mdr-1), and breast cancer resistant protein (BCRP) that are critical in efflux of harmful hydrophilic and hydrophobic xenobiotics from the brain parenchyma [76-78]. In addition, however, they also serve to efflux many pharmaceutical compounds, limiting their therapeutic value.
Low transcellular transport at the BBB is governed by low numbers of caveolae and reduced transcellular transporters such as major facilitator superfamily domain-containing protein -2 (MFSD2 or MFSD2A) [79,80]. Specifically, MFSD2A is an DHA omega3 fatty acid transporter in endothelial cells where it regulates vesicular traffic in CNS BBB/blood retinal barrier (BRB) [81,82]. Systemic ablation of MFSD2A increased BBB permeability due to uncontrolled vesicular trafficking in endothelial cells [83]. Caveolin-1 regulates signal transduction, endocytosis, transcytosis and molecular transport. It also controls angiogenic response through mediating VEGF receptor 2 (VEGFR2) phosphorylation and internalization [49,70,84]. Caveolin-1; however, contributes to junctional opening by weakening VE-cadherin based adherens junction by interacting with β-catenin[85]. Under inflammatory conditions, the pro inflammatory chemokine CCL2 induces BBB disruption through CAV-1 mediated internalization of occludin and claudin-5 [86].
Enzymes that make chemical modifications to molecules that may cross the BBB and affect neuronal function are also prevalent in brain endothelial cells. These chemical modifications reduce toxicity of molecules by either metabolism that renders the molecules inactive or by addition of moieties that directs the toxins towards excretory pathways [87,88]. Specifically, brain endothelial cells have high concentrations of γ-glutamyl transpeptidase, alkaline phosphatase and aromatic acid decarboxylases [89]. Together with active transporters, these metabolic barriers regulates the concentrations of ions, metabolites and foreign substances within the NVU [74,90].
Pericytes
Pericytes wrap around the small vessels thereby providing both structural and vasodynamic support to the microvasculature. Pericytes are involved in cerebral autoregulation through their expression of receptors that are critical for transmitters such as catecholamines [91], angiotensin II [92], vasoactive intestinal peptides [93], endothelin-1 [94], and vasopressin [95]. Ramsauer and colleagues have shown that pericytes stabilize the capillary like structures that are formed by endothelial cells during angiogenesis and differentiation of the BBB [96]. Furthermore, the use of viable pericyte deficient mouse mutants have shown that pericyte deficiency increases BBB permeability to water and to a range of low to high molecular weight tracers [46]. This increased permeability has been shown to occur through endothelial transcytosis indicating that pericyte regulation of the transcellular barrier is critical to the normal function of the BBB [97]. Other conditional knockout mouse models have also shown that loss of pericytic laminin causes BBB breakdown and hydrocephalus [98]. This can be attributed to the destabilization of the basement membrane that normally promotes BBB integrity by providing structural support for the endothelial cells, pericytes and astrocytes. Diminished expression of critical transporters such as aquaporin 4 (AQP4) in conditional knockouts is also linked to hydrocephalus [98]. BBB-specific gene expression patterns in endothelial cells and induction of polarization in astrocyte foot processes are also regulated by pericytes [46].
Basement membrane
The NVU basement membrane contributes to the integrity of the BBB by providing structural support for both endothelial cells and pericytes. The cerebrovascular basement membrane mainly consists of extracellular matrix proteins; collagen IV, laminin, perlecan, nidogen and fibronectin that are released by endothelial cells, pericytes and astrocytes [99-101]. Notably, the neurovascular unit consists of two types of the basement membrane, (BL1 Figure 3) the endothelial basement membrane formed by endothelial cells and pericytes and (BL2 Figure 3) the parenchymal basement membrane that consist of extra cellular matrix proteins secreted by astrocytes [102-105]. The protein composition in these perivascular spaces are distinctly different; the endothelial basement membrane consists of laminin α4 and α5 [106] while the parenchymal membrane is composed of laminin α1 and α2 [102,103]. This particular protein composition contributes to specific signaling within the NVU that is necessary for normal function of the BBB. Hence, the basement membrane serves not only as a physical barrier but also regulates signaling pathways in endothelial cells and pericytes thereby influencing BBB integrity [102,107,108].
Many current three-dimensional models consist of artificial polycarbonate, polytetrafluorethylene, and polydimethyl-siloxane membranes serving as basal lamina [5,30,109], which is not reflective of the human brain ECM. Even though the use of collagen (usually type I) to provide a 3D microenvironment for glial cells and neurons [5] allows for the fabrication of multi cellular in-vitro models, it is important to note that no collagen I and very low amounts of collagen type 4 are found in the adult human brain. The ECM in the adult brain tissue consists of lecticans, a family of proteoglycans that contain lectin and hyaluronic acid domains [110]. Incorporation of such ECM may promote BBB maturation that recapitulates normal human physiology.
Many current three-dimensional models consist of artificial polycarbonate, polytetrafluorethylene, and polydimethyl-siloxane membranes serving as basal lamina [5,30,109], which is not reflective of the human brain ECM. Even though the use of collagen (usually type I) to provide a 3D microenvironment for glial cells and neurons [5] allows for the fabrication of multi cellular in-vitro models, it is important to note that no collagen I and very low amounts of collagen type 4 are found in the adult human brain. The ECM in the adult brain tissue consists of lecticans, a family of proteoglycans that contain lectin and hyaluronic acid domains [110]. Incorporation of such ECM may promote BBB maturation that recapitulates normal human physiology.
Astrocytes
Astrocytes are the most abundant cell type in the vertebrate CNS. They have specialized end feet covering the surface of the CNS microvessels [49], providing a close association to the microvessels. Astrocytes express crucial proteins such as aquaporin-4 (AQP-4) and Kir4 potassium channels at the foot processes that function to regulate water homeostasis in the NVU [111]. In vitro experiments demonstrated that astrocytes improved BBB integrity in co-culture models with brain endothelial cells and administrating astrocyte conditioned media to the brain endothelial cells also improved BBB integrity signifying that astrocytes secrete soluble factors that enhance BBB integrity [112-115]. Specifically, astrocytes improve and maintain BBB integrity through the secretion of factors Wnts and norrin [116], and also release sonic hedgehog, retinoic acid, and angiopotin-1, which are key factors that support barrier properties in brain endothelial cells [117-119]. The crosstalk between endothelial cells and the astrocytes is not only critical for improving and maintaining the BBB integrity but it is also vital for cellular differentiation that promotes BBB maturation. Specifically, endothelial cells secrete leukemia inhibitory factor-1 that supports astrocyte differentiation that in turn induces the expression of Src suppressed C-Kinase substrate, leading to astrocytic secretion of angiopotin-1, which stabilizes vessels through Tie2/TEK binding on endothelial cells [111].
Microglia
As a distinct class of the glial cells, microglia serve as the primary brain immune effector cells that become activated and undergo morphological and functional transformation during various brain injuries and diseases [90,120]. They are ontogenetically related to the mononuclear phagocyte lineage and are activated by lesions, neurodegenerative diseases, stroke, and brain neoplasm [90,121-124]. Microglial response at a site of injury are complex, however, structural changes such as motile branches and migration of stomata have been observed during microglial response to insults [125,126]. In their resting phase, microglia have long, thin ramified processes [120]. During this phase, microglia are in a vigilant form because they can promptly sense slight homeostatic disturbance in the CNS. The changes from the resting phase to the activated form is antigen specific and orchestrated by specific cytokine activation [75,127-129].
Microglial involvement in brain injuries has been investigated but there are very few studies that have been conducted to delineate a direct link between microglia and BBB maintenance. Glial involvement in BBB maintenance has mostly been attributed to astrocytes because their end feet processes touch the microvessels. Findings on microglial location in the perivascular space highlight their interaction with endothelial cells and supports their influence on BBB integrity. During their resting phase, microglial cells effectively control the neurovascular unit microenvironment. Nimmerjahn and colleagues, have shown that microglia clear the parenchyma of accumulated low diffusible metabolic products and tissue component debris [120]. They observed bulbous branch endings and spontaneous engulfment of tissue components. Further histologic staining highlighted microglial processes and protrusions in contact with neuronal cell bodies and blood vessels [120]. This indicates that under healthy conditions, microglia interact with other cortical elements and regulate the NVU microenvironment by clearing debris and cellular components. However, it should be noted that upon activation, microglia release cytokines such as TNF-α and IL-6 that have been associated with BBB dysfunction [130,131]. Further discussion on microglia involvement in disease states will be included below in specific disease sections of this report. However, more studies are needed to elucidate the direct interaction between microglia and cells of the BBB in order to fully understand the involvement of microglia in BBB maintenance. Current in vitro neurovascular unit models do not consist of microglia, in contrast to our current model.
Microglial involvement in brain injuries has been investigated but there are very few studies that have been conducted to delineate a direct link between microglia and BBB maintenance. Glial involvement in BBB maintenance has mostly been attributed to astrocytes because their end feet processes touch the microvessels. Findings on microglial location in the perivascular space highlight their interaction with endothelial cells and supports their influence on BBB integrity. During their resting phase, microglial cells effectively control the neurovascular unit microenvironment. Nimmerjahn and colleagues, have shown that microglia clear the parenchyma of accumulated low diffusible metabolic products and tissue component debris [120]. They observed bulbous branch endings and spontaneous engulfment of tissue components. Further histologic staining highlighted microglial processes and protrusions in contact with neuronal cell bodies and blood vessels [120]. This indicates that under healthy conditions, microglia interact with other cortical elements and regulate the NVU microenvironment by clearing debris and cellular components. However, it should be noted that upon activation, microglia release cytokines such as TNF-α and IL-6 that have been associated with BBB dysfunction [130,131]. Further discussion on microglia involvement in disease states will be included below in specific disease sections of this report. However, more studies are needed to elucidate the direct interaction between microglia and cells of the BBB in order to fully understand the involvement of microglia in BBB maintenance. Current in vitro neurovascular unit models do not consist of microglia, in contrast to our current model.
Oligodendrocytes
Oligodendrocytes, like Schwann cells in the peripheral nervous system, are glial cells responsible for the formation of myelin sheets that provide support and insulation to axons in the central nervous system [90]. Oligodendrocytes produce a myelin sheet membrane consisting of lipids and specialized proteins that wraps around the axons in the CNS [90] and provides vital support for proper electrical signal transmission. Since a single oligodendrocyte can extend its processes to about 50 axons [132], the effects of oligodendrocyte dysfunction can be deleterious. More importantly, oligodendrocytes are known to secret TGF-β, a factor that promotes BBB integrity through the activation of the MEK/ERK pathway, which consequently upregulates expression of tight junction proteins for BBB maturation and maintenance [36]. Rhodes and colleagues have shown that lesions or injuries that results in oligodendrocytes dysfunction disrupts the BBB [133]. No definitive studies have been conducted to determine whether oligodendrocyte dysfunction causes BBB breakdown or whether it is the breakdown of the BBB that results in oligodendrocyte dysfunction that consequently leads to neuronal dysfunction. Rhodes and colleagues showed that BBB breakdown using VEGF or lipopolysaccharide in rats and mice caused hypertrophy in oligodendrocytes precursor cells. Direct injection of blood components such as serum, thrombin did not have an effect. However, platelets, macrophages, TGF-β, TNF-α and IL-1 caused hypertrophy and increased NG2 in oligodendrocyte precursor cells [133]. Further studies that incorporate diseased oligodendrocyte or other parenchymal cells in animal or in vitro models may help delineate the effect of dysfunctional oligodendrocytes on the BBB integrity. Most existing in vitro models do not include oligodendrocytes and hence their contribution to BBB maturation is not appropriately modeled. We incorporated oligodendrocyte precursor cells in our model to more closely mimic normal human physiology of the neurovascular unit.
Neurons
The concerted interaction between neurons, glial cells and endothelial cells termed neurovascular coupling, determines BBB formation and function in vertebrates. As depicted in (Figure 1), astrocyte processes enwrap synaptic terminals, which allows them to transmit signals from neurons to the microvessels [41,134]. However, the mechanisms of neuronal involvement in BBB maturation and maintenance is not well understood. Co-culture of rat brain endothelial cells with differentiated neuro progenitor cells (NPCs) revealed some clues that neurons influence BBB integrity [135]. Neuro-endothelial cell co-culture studies have shown that neurons increase trans-endothelial electrical resistance and decreases permeability in endothelial cells [135,136]. Additionally, Lippmann’s results from co-cultures of endothelial cells with NPCs showed upregulation of 10 endothelial cell genes [135]. Some of the genes that were upregulated included genes that induce angiogenesis, and CPE, a gene that encodes for carboxypeptidase E, an enzyme that regulates brain derived neurotrophic factor processing [135]. The upregulation of the CPE gene in endothelial cells indicates an increase of the peptide reflecting the cross talk between neurons and endothelial cells. They also showed that neurons increased MDR1A expression, a gene that encodes for a critical efflux transporter at the BBB [135]. Those studies indicate that increasing the complexity of co-culture models to include multiple neural cells instead of just primary BBB cells increased not only BBB integrity, but may also help create platforms that can be utilized in many applications beyond BBB studies.
BBB DYSFUNCTION IN NEURODEGENERATIVE DISEASES
Mechanistic causes for most neurological disorders are not known, however, blood-brain barrier dysfunction and inflammation play major roles in neurodegenerative disease pathologies [75,137-143]. To highlight major points BBB malfunction associated in neurological disorders, the mechanism of BBB breakdown in Alzheimer’s disease, Parkinson’s disease and multiple sclerosis will be presented. It should be noted, however, that BBB malfunction in Amyotrophic Lateral Sclerosis (ALS) [144-146], Huntington’s disease [147-149], HIV associated dementia [150-153] and other neurological diseases not described here also contribute to those disease pathologies.
Alzheimer’s disease
Alzheimer’s disease (AD) is progressive degenerative brain disorder that is characterized by the accumulation of plaques in the brain, irreversible cognitive impairment, decline in thinking and memory and thus a decline in behavioral and social skills. Characterized by an accumulation of protein plaques and tangles in the brain, it is the most common cause of adult dementia. Schneck and colleagues compiled the current concepts in AD [154], so this discussion will be limited to BBB dysfunction in AD.
Over 20 independent postmortem studies have confirmed BBB breakdown in AD [155]. Hallmarks of BBB dysfunction include, pericytes and endothelial cell degeneration, loss of tight junctions, red blood cell and monocyte extravasation, brain capillary leakages of blood borne components such as albumin, immunoglobulin (IgG), fibrinogen, and thrombin [139]. There are many mechanisms that contribute to BBB breakdown in AD. Some of these processes include early cerebrovascular disorder [156], vascular dysregulation [157], and ischemic damage [158].
Though many causes of AD have not been linked to specific genetic causes, transgenic animal models are prevalently used to study Alzheimer’s disease [122,159-162]. Mice with mutations in the APP gene, which produces the amyloid precursor protein that is in turn processed into fragments including amyloid beta (Aβ) peptide, have been shown to have capillary leakages of blood derived fibrinogen, IgG and albumin, and leakage of experimentally injected Evans blue dyes. Electron microscopy of these mouse brains also indicated degeneration and loss of pericytes, endothelium, and vascular smooth muscles cells [163-167]. Time course studies evaluating BBB breakdown, with respect to other pathologies, indicated that BBB breakdown develops early in APP transgenic mice [52,166-168]. Deane and colleagues observed aberrant expression of low-density lipoprotein transporter protein1 (LRP1) in APP transgenic mice [169]. This transporter is a major efflux protein for Aβ toxin at the BBB. Other studies with an APP model showed an increase in expression of an influx transporter, the receptor for advanced glycation end products (RAGE) [170]. Normal function of transporters such as LRP1 and RAGE at the BBB is crucial for maintaining brain homeostasis.
Cerebrovascular autoregulation is impaired in APP murine models [171]. For example, studies have shown reduced brain glucose uptake due to glucose transporter dysfunction in APP transgenic models[171,172]. BBB breakdown and inhibition of LRP1 transcription due to diminished GLUT1 expression in brain endothelial cells is thought to accelerate Aβ pathology [172]. Alterations in protein expression of all these crucial transporters at the BBB can result in a malfunctioning BBB that can lead to deleterious secondary consequences.
BBB breakdown has been shown in many other transgenic murine models representing known or suspected genetic associations from human studies. For examples, loss of BBB integrity [173] and loss of vascular phenotype [164] in mice expressing PSEN1 mutation, BBB breakdown in Tau transgenic mice [174], Pericyte degeneration and BBB dysfunction in PDGFRβ- deficient transgenic mice [26], and accumulation of perivascular IgG, fibrinogen, thrombin, hemosiderin deposits and leakage of Evans blue in APOE transgenic mice [175]. Even though no effective treatment has been developed from any of these animal models, it is worth noting that the models have increased our understanding of molecular pathways in brain cellular functions and may possibly help to identify biomarkers for early Alzheimer’s disease diagnosis.
Importantly, neuroimaging in patients with mild cognitive impairment revealed that BBB breakdown precedes brain atrophy or dementia [168,176-178]. Further studies and new models are still needed to determine cellular and molecular mechanism by which the BBB is impaired and to accelerate the development of therapeutic targets for Alzheimer’s disease that aim to maintain and repair BBB integrity.
Over 20 independent postmortem studies have confirmed BBB breakdown in AD [155]. Hallmarks of BBB dysfunction include, pericytes and endothelial cell degeneration, loss of tight junctions, red blood cell and monocyte extravasation, brain capillary leakages of blood borne components such as albumin, immunoglobulin (IgG), fibrinogen, and thrombin [139]. There are many mechanisms that contribute to BBB breakdown in AD. Some of these processes include early cerebrovascular disorder [156], vascular dysregulation [157], and ischemic damage [158].
Though many causes of AD have not been linked to specific genetic causes, transgenic animal models are prevalently used to study Alzheimer’s disease [122,159-162]. Mice with mutations in the APP gene, which produces the amyloid precursor protein that is in turn processed into fragments including amyloid beta (Aβ) peptide, have been shown to have capillary leakages of blood derived fibrinogen, IgG and albumin, and leakage of experimentally injected Evans blue dyes. Electron microscopy of these mouse brains also indicated degeneration and loss of pericytes, endothelium, and vascular smooth muscles cells [163-167]. Time course studies evaluating BBB breakdown, with respect to other pathologies, indicated that BBB breakdown develops early in APP transgenic mice [52,166-168]. Deane and colleagues observed aberrant expression of low-density lipoprotein transporter protein1 (LRP1) in APP transgenic mice [169]. This transporter is a major efflux protein for Aβ toxin at the BBB. Other studies with an APP model showed an increase in expression of an influx transporter, the receptor for advanced glycation end products (RAGE) [170]. Normal function of transporters such as LRP1 and RAGE at the BBB is crucial for maintaining brain homeostasis.
Cerebrovascular autoregulation is impaired in APP murine models [171]. For example, studies have shown reduced brain glucose uptake due to glucose transporter dysfunction in APP transgenic models[171,172]. BBB breakdown and inhibition of LRP1 transcription due to diminished GLUT1 expression in brain endothelial cells is thought to accelerate Aβ pathology [172]. Alterations in protein expression of all these crucial transporters at the BBB can result in a malfunctioning BBB that can lead to deleterious secondary consequences.
BBB breakdown has been shown in many other transgenic murine models representing known or suspected genetic associations from human studies. For examples, loss of BBB integrity [173] and loss of vascular phenotype [164] in mice expressing PSEN1 mutation, BBB breakdown in Tau transgenic mice [174], Pericyte degeneration and BBB dysfunction in PDGFRβ- deficient transgenic mice [26], and accumulation of perivascular IgG, fibrinogen, thrombin, hemosiderin deposits and leakage of Evans blue in APOE transgenic mice [175]. Even though no effective treatment has been developed from any of these animal models, it is worth noting that the models have increased our understanding of molecular pathways in brain cellular functions and may possibly help to identify biomarkers for early Alzheimer’s disease diagnosis.
Importantly, neuroimaging in patients with mild cognitive impairment revealed that BBB breakdown precedes brain atrophy or dementia [168,176-178]. Further studies and new models are still needed to determine cellular and molecular mechanism by which the BBB is impaired and to accelerate the development of therapeutic targets for Alzheimer’s disease that aim to maintain and repair BBB integrity.
Parkinison’s disease
Parkinson’s disease (PD) is a progressive neurodegenerative disorder that is characterized by neuronal death in the substantia nigra basal ganglia, degeneration of dopaminergic neurotransmission, and the presence of Lewy body (α-synuclein) protein deposits [179-181]. PD symptoms include asymmetrical bradykinesia, rigidity, resting tremor and postural instability [182,183]. Cellular events such as failure in the protein degradation machinery, oxidative stress, mitochondrial dysfunction, defects in mitophagy and the accumulation of α-synuclein are believed to drive PD initiation and progression [180,184,185]. Vascular damage from α-synuclein deposition increases BBB permeability which suggests the role of the protein in BBB disruption and PD development [180,186,187]. These and other cellular dysfunctions lead to glial or neuronal cell death.
While some studies in animal models assume that BBB integrity remains unchanged during the development of PD pathology [188,189], clinical evidence shows increased BBB permeability in PD patients [190-193]. PD patients have been shown to have reduced P-glycoprotein (P-gp) function [191,194]. Studies in P-gp knock-out mice have shown an increased parenchymal accumulation of administered neurotoxins, ivermectin and the carcinostatic vinblanstine. Hence normal P-gp function at the BBB appears compromised in PD [195]. Furthermore, diminished P-gp activity in aged people is associated with reduced removal of toxins from the brain and linked to PD pathology [196]. Since PD is a chronic neurodegenerative disorder that affects one in every 100 people at the age of 60 and above, it is worth pointing out that many age related processes, such as increased production of ROS and proinflammatory cytokines in brain endothelial cells, contribute heavily towards BBB dysfunction [196-198]. Since neurons and other parenchymal cells are mainly affected in PD and other age-related dementias, it is critical that in vitro models designed to understand molecular involvement of the BBB in PD contain these cell types.
While some studies in animal models assume that BBB integrity remains unchanged during the development of PD pathology [188,189], clinical evidence shows increased BBB permeability in PD patients [190-193]. PD patients have been shown to have reduced P-glycoprotein (P-gp) function [191,194]. Studies in P-gp knock-out mice have shown an increased parenchymal accumulation of administered neurotoxins, ivermectin and the carcinostatic vinblanstine. Hence normal P-gp function at the BBB appears compromised in PD [195]. Furthermore, diminished P-gp activity in aged people is associated with reduced removal of toxins from the brain and linked to PD pathology [196]. Since PD is a chronic neurodegenerative disorder that affects one in every 100 people at the age of 60 and above, it is worth pointing out that many age related processes, such as increased production of ROS and proinflammatory cytokines in brain endothelial cells, contribute heavily towards BBB dysfunction [196-198]. Since neurons and other parenchymal cells are mainly affected in PD and other age-related dementias, it is critical that in vitro models designed to understand molecular involvement of the BBB in PD contain these cell types.
Multiple sclerosis
Multiple sclerosis (MS) is a chronic autoimmune, inflammatory neurological disease that is characterized by demyelinating plaques in the central nervous system (CNS) [199-201]. MS symptoms include visual disturbances, numbness, prickling, muscle weakness, loss of coordination and balance, thinking and memory deficits [202]. The disease starts to manifest between the ages of 20 and 40. The cause of MS is unknown and there is no cure for the disease. The only existing therapies are based on blocking transmigration of T cells across the BBB [203]. Leukocyte entry into the CNS is one of the hallmarks of MS. Activated leukocytes, specifically autoaggressive CD4+ T lymphocytes, are believed to accumulate in the brain by traversing the BBB and the cerebral spinal fluid (CSF) barrier through steps including, rolling, activation, adhesion and transmigration [204-208]. Chemokines activate integrins on leukocytes to enhance adhesion. This binding subsequently leads to cytoskeletal reorganization of G-protein-coupled receptors, which allows the transmigration of leukocytes [205,209].
The transmigration of leukocytes highlights the importance of maintaining BBB integrity in MS. Neuroimaging studies and postmortem findings in MS patients show that BBB disruption is an early feature in MS [83] and animal studies have shown that BBB breakdown precedes leukocyte infiltration [47,83]. Spencer and colleagues speculated that environmental and genetic associations may influence the BBB, which results in the vessel pathology of the disease [201]. In order to elucidate the mechanistic involvement of the BBB in MS pathology, new in vitro models that contain vascular cells and neuro-glial components such as oligodendrocytes and neurons are critical to model a functional BBB. Such models can be applied to assess transmigration of leukocytes. Models containing a functional BBB can be used to identify disease initiating microenvironmental factors, such as changes in cytokine levels, that activate circulating leukocytes and initiate adhesion. Further, such models could be utilized to investigate pathological connections between BBB integrity and demyelination that occurs in MS. Current in vitro models of the NVU do not contain oligodendrocytes or neuronal cells types, which makes it difficult to study MS disease conditions comprehensively in vitro.
The transmigration of leukocytes highlights the importance of maintaining BBB integrity in MS. Neuroimaging studies and postmortem findings in MS patients show that BBB disruption is an early feature in MS [83] and animal studies have shown that BBB breakdown precedes leukocyte infiltration [47,83]. Spencer and colleagues speculated that environmental and genetic associations may influence the BBB, which results in the vessel pathology of the disease [201]. In order to elucidate the mechanistic involvement of the BBB in MS pathology, new in vitro models that contain vascular cells and neuro-glial components such as oligodendrocytes and neurons are critical to model a functional BBB. Such models can be applied to assess transmigration of leukocytes. Models containing a functional BBB can be used to identify disease initiating microenvironmental factors, such as changes in cytokine levels, that activate circulating leukocytes and initiate adhesion. Further, such models could be utilized to investigate pathological connections between BBB integrity and demyelination that occurs in MS. Current in vitro models of the NVU do not contain oligodendrocytes or neuronal cells types, which makes it difficult to study MS disease conditions comprehensively in vitro.
NVU CONTAINING A FUNCTIONAL BBB
Most current NVU models are not integrated with both glia and neuronal components and are limited in addressing scientific enquiries concerning the link between BBB dysfunction in neurological disorders and the parenchymal tissue components, or the effects of drug candidates after they cross the BBB. Our lab developed a six cell type brain neurovascular unit organoid model that produces a functional BBB[210]. The organoid model contains human brain microvascular endothelial cells (HE), human pericytes (HP), human astrocytes (HA), human microglia (HM), human oligodendrocytes (HO) and human neurons (HN), with surrounding endothelial cells enclosing the brain parenchymal cells. We reported cell viability for up to 21 days in vitro that could be critical in assessing the long-term effect in drug toxicity studies. We also reported the expression of the efflux protein, P-gp that exported xenobiotics from the brain tissue. Importantly, our data show that Hg2+, from HgCl2 dissolved in the media, only causes significant cell death in neuronal organoids that did not have a BBB (no endothelial cells or pericytes) compared to organoids that possess an intact BBB. This indicated charge selectivity in the organoid model containing all 6 cell types. This finding was further validated using a small molecule pro-drug MPTP (a lipophilic small molecule), which is toxic when enzymatically converted into its positively charged metabolite MPP+ in glial cells. MPTP caused low ATP production and cell death in organoids that contained all six cell types and an intact BBB. In contrast, charged MPP+ did not cause cell death to organoids containing 6 cell types, but significantly decreased ATP production to organoids without an intact BBB [210]. These toxicity studies reflect the normal function of the BBB that is critical in maintaining brain homeostasis.
We have highlighted above that BBB breakdown precedes brain atrophy and leucocyte infiltration in Alzheimer’s and MS respectively. Upon investigating BBB integrity, our data from this organoid model show transmigration of 70 kDa dextran and IgG when the BBB is transiently disrupted using histamine or hypoxia. Further studies are needed to assess the link between BBB leakage and neurological disorders. Current APOE transgenic mice models also show accumulation of perivascular IgG, fibrinogen, thrombin, hemosiderin deposits and leakage of Evans blue into the brain parenchyma. A model containing functional BBB and neuro-glia components may help elucidate the link between BBB disruption and Alzheimer’s disease and related dementias. Our data also show the disruption of tight junctions when the organoids are cultured under hypoxic conditions. This indicates the utility of the model in understanding the underlying physiological conditions that alter normal NVU microenvironment that could possibly cause BBB breakdown in neurological disorders. Further evaluation of this model is required to ascertain its utility in neurodegenerative disease modeling.
In this review we have described the importance of the integrated function of the cell types that make up the BBB with parenchymal cells such as glia and neurons. We have discussed the limitation of 2D models that do not recapitulate the basic functions of the BBB. 2D cell cultures are limited in disease modeling applications because they do not allow for the incorporation of more than 3 cell types without losing the proper 3D microenvironment required to recapitulate normal physiological function of each of the cell types. We also highlighted the fact that most current models incorporate three major cell types that form the BBB or use rodent cells to imply human BBB physiology and functionality. We propose that the use of these models in preclinical studies may not fully represent normal NVU physiology activity. Specifically, receptor and enzyme systems that regulate the influx and efflux of substances at the BBB level are different between species and hence extrapolation of BBB functionality from rodents needs to be done with caution and/or with a fair understanding of these differences. For example, studies have shown that some radio ligands that are substrates for P-gp in rodents are efficiently exported in rodents [211]. Similar studies, however, have shown that these radio ligands are taken up and retained by the brain in humans and non-human primates [212-214]. Furthermore, we have also stated that the utilization of synthetic membranes to establish the basement membrane is limiting for they impede important intercellular interactions that are critical for the normal function of the neurovascular unit to maintain BBB integrity. However, new models consisting of electro spun membranes derived from human ECM may enhance the development of physiologically relevant NVU model.
We have highlighted above that BBB breakdown precedes brain atrophy and leucocyte infiltration in Alzheimer’s and MS respectively. Upon investigating BBB integrity, our data from this organoid model show transmigration of 70 kDa dextran and IgG when the BBB is transiently disrupted using histamine or hypoxia. Further studies are needed to assess the link between BBB leakage and neurological disorders. Current APOE transgenic mice models also show accumulation of perivascular IgG, fibrinogen, thrombin, hemosiderin deposits and leakage of Evans blue into the brain parenchyma. A model containing functional BBB and neuro-glia components may help elucidate the link between BBB disruption and Alzheimer’s disease and related dementias. Our data also show the disruption of tight junctions when the organoids are cultured under hypoxic conditions. This indicates the utility of the model in understanding the underlying physiological conditions that alter normal NVU microenvironment that could possibly cause BBB breakdown in neurological disorders. Further evaluation of this model is required to ascertain its utility in neurodegenerative disease modeling.
In this review we have described the importance of the integrated function of the cell types that make up the BBB with parenchymal cells such as glia and neurons. We have discussed the limitation of 2D models that do not recapitulate the basic functions of the BBB. 2D cell cultures are limited in disease modeling applications because they do not allow for the incorporation of more than 3 cell types without losing the proper 3D microenvironment required to recapitulate normal physiological function of each of the cell types. We also highlighted the fact that most current models incorporate three major cell types that form the BBB or use rodent cells to imply human BBB physiology and functionality. We propose that the use of these models in preclinical studies may not fully represent normal NVU physiology activity. Specifically, receptor and enzyme systems that regulate the influx and efflux of substances at the BBB level are different between species and hence extrapolation of BBB functionality from rodents needs to be done with caution and/or with a fair understanding of these differences. For example, studies have shown that some radio ligands that are substrates for P-gp in rodents are efficiently exported in rodents [211]. Similar studies, however, have shown that these radio ligands are taken up and retained by the brain in humans and non-human primates [212-214]. Furthermore, we have also stated that the utilization of synthetic membranes to establish the basement membrane is limiting for they impede important intercellular interactions that are critical for the normal function of the neurovascular unit to maintain BBB integrity. However, new models consisting of electro spun membranes derived from human ECM may enhance the development of physiologically relevant NVU model.
FUTURE PERSPECTIVES
There is a great need for complex in vitro models that can be utilized in pre-clinical studies including drug discovery, toxicity screening, and biomarker identification studies. Since the model we have developed has only been evaluated for BBB permeability and the effect of hypoxia on BBB function, it should be noted that further work is required to ascertain the utility of the model in pre-clinical studies. To do this, one will have to establish the transport pathways present at the BBB in our organoid system. Such understanding would pave the way for developing strategies by which drug candidates could be structurally and chemically optimized to enhance permeability across the BBB without compromising BBB integrity.
Treatment schemes that are best suited for an individual based on pharmacogenetic and pharmacogenomic information have been reported for cancer [215-218]. This theme of personalized medicine could prove to be very effective in genetically influenced neurological diseases [219-221]. To pave the way for extensive studies, in vitro models containing patient derived cells could be utilized not only to identify therapeutic targets that are specific to the individual or group of individuals that express specific genotypes but would also aid the understanding of molecular and biochemical basis of drug efficacy. Future studies incorporating patient derived cells into an organoid system containing the major components of the NVU could elucidate mechanistic connections between BBB dysfunction and disease progression. Patient derived cells of all cell types composing the NVU are not readily available. In addition, efficient methods of deriving all six cell types composing the NVU through the use of induced pluripotent stem cell (iPSC) technology have not been developed to date. To overcome this limitation, gene editing tools could be employed to create mutations in specific cell types prior to incorporating them into an organoid. The organoid could then be evaluated for that disease phenotype.
Treatment schemes that are best suited for an individual based on pharmacogenetic and pharmacogenomic information have been reported for cancer [215-218]. This theme of personalized medicine could prove to be very effective in genetically influenced neurological diseases [219-221]. To pave the way for extensive studies, in vitro models containing patient derived cells could be utilized not only to identify therapeutic targets that are specific to the individual or group of individuals that express specific genotypes but would also aid the understanding of molecular and biochemical basis of drug efficacy. Future studies incorporating patient derived cells into an organoid system containing the major components of the NVU could elucidate mechanistic connections between BBB dysfunction and disease progression. Patient derived cells of all cell types composing the NVU are not readily available. In addition, efficient methods of deriving all six cell types composing the NVU through the use of induced pluripotent stem cell (iPSC) technology have not been developed to date. To overcome this limitation, gene editing tools could be employed to create mutations in specific cell types prior to incorporating them into an organoid. The organoid could then be evaluated for that disease phenotype.
SUMMARY
Complex models containing all the cellular components of the NVU may aid, drug discovery, neurotoxicity screening, biomarker identification and the development of new therapies for neurological disorders. More importantly, human cell-derived complex models containing functional BBB, glia and neurons will help select drug candidates that can not only cross the BBB but will also help understand the effects of the molecules after they cross the BBB. This is important not only for drug candidates against neurological disorders but even for evaluating toxicity effects of brain penetrating drugs and metabolites that are administered systematically.
ACKNOWLEDGEMENT
This work was supported by a grant to Wake Forest Institute for Regenerative Medicine from the State of North Carolina (# -117249).
REFERENCES
- Hirtz D, Thurman DJ, Gwinn-Hardy K, Mohamed M, Chaudhuri AR, et al. (2007) How common are the “common” neurologic disorders? Neurology 68: 326-337.
- Alzheimer’s Association (2015) 2015 Alzheimer's disease facts and figures. Alzheimer's Dement 11: 332-384.
- Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J (2014) Clinical development success rates for investigational drugs. Nat Biotechnol 32: 40-51.
- Bowman PD, Ennis SR, Rarey KE, Betz AL, Goldstein GW (1983) Brain microvessel endothelial cells in tissue culture: A model for study of blood?brain barrier permeability. Ann Neurol 14: 396-402.
- Brown JA, Pensabene V, Markov DA, Allwardt V, Neely MD, et al. (2015) Recreating blood-brain barrier physiology and structure on chip: A novel neurovascular microfluidic bioreactor. Biomicrofluidics 9: 054124.
- Cecchelli R, Berezowski V, Lundquist S, Culot M, Renftel M, et al. (2007) Modelling of the blood-brain barrier in drug discovery and development. Nat Rev Drug Discov 6: 650-661.
- Cho CF, Wolfe JM, Fadzen CM, Calligaris D, Hornburg K, et al. (2017) Blood-brain-barrier spheroids as an in vitro screening platform for brain-penetrating agents. Nat Commun 8: 15623.
- Cucullo L, Aumayr B, Rapp E, Janigro D (2005) Drug delivery and in vitro models of the blood-brain barrier. Curr Opin Drug Discov Devel 8: 89-99.
- Deosarkar SP, Prabhakarpandian B, Wang B, Sheffield JB, Krynska B, et al. (2015) A novel dynamic neonatal blood-brain barrier on a chip. PLoS One 10: e0142725.
- Freese C, Reinhardt S, Hefner G, Unger RE, Kirkpatrick CJ, et al. (2014) A novel blood-brain barrier co-culture system for drug targeting of alzheimer’s disease: Establishment by using acitretin as a model drug. PloS One 9: e91003.
- Helms HC, Abbott NJ, Burek M, Cecchelli R, Couraud PO, et al. (2016) In vitro models of the blood–brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. J Cereb Blood Flow Metab 36: 862-890.
- Janigro D, Strelow L, Grant G, Nelson JA (1998) In vitro blood-brain barrier model for HIV-induced CNS disease. Neuro AIDS 1.
- Phan DT, Bender RHF, Andrejecsk JW, Sobrino A, Hachey SJ, et al. (2017) Blood-brain barrier-on-a-chip: Microphysiological systems that capture the complexity of the blood–central nervous system interface. Exp Biol Med (Maywood) 242: 1669-1678.
- Poller B, Gutmann H, Krähenbühl S, Weksler B, Romero I, et al. (2008) The human brain endothelial cell line hCMEC/D3 as a human blood?brain barrier model for drug transport studies. J Neurochem 107: 1358-1368.
- Prieto P, Blaauboer BJ, de Boer AG, Boveri M, Cecchelli R, et al. (2004) Blood-brain barrier in vitro models and their application in toxicology. Altern Lab Anim 32: 37-50.
- Rubin LL, Hall DE, Porter S, Barbu K, Cannon C, et al. (1991) A cell culture model of the blood-brain barrier. J Cell Biol 115: 1725-1735.
- Wang JD, Khafagy el-S, Khanafer K, Takayama S, ElSayed ME (2016) Organization of endothelial cells, pericytes, and astrocytes into a 3d microfluidic in vitro model of the blood-brain barrier. Mol Pharm 13: 895-906.
- Wilhelm I, Fazakas C, Krizbai IA (2011) In vitro models of the blood-brain barrier. Acta Neurobiol Exp 71: 113-128.
- Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, et al. (2013) Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A 110: 3507-3512.
- Arai K, Lo EH (2009) An oligovascular niche: Cerebral endothelial cells promote the survival and proliferation of oligodendrocyte precursor cells. J Neurosci 29: 4351-4355.
- Das A, Frank RN, Weber ML, Kennedy A, Reidy CA, et al. (1988) ATP causes retinal pericytes to contract in vitro. Exp Eye Res 46: 349-362.
- Raza A, Franklin MJ, Dudek AZ (2010) Pericytes and vessel maturation during tumor angiogenesis and metastasis. Am J Hematol 85: 593-598.
- Cohen MP, Frank RN, Khalifa AA (1980) Collagen production by cultured retinal capillary pericytes. Invest Ophthalmol Vis Sci 19: 90-94.
- Hellstrom M,Gerhardt H, Kalén M, Li X, Eriksson U, et al. (2001) Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol 153: 543-553.
- Armulik A, Mäe M, Betsholtz C (2011) Pericytes and the blood-brain barrier: recent advances and implications for the delivery of CNS therapy. Ther Deliv 2: 419-422.
- Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, et al. (2010) Pericytes regulate the blood-brain barrier. Nature 468: 557-561.
- Beck DW, Roberts RL, Olson JJ (1986) Glial cells influence membrane-associated enzyme activity at the blood-brain barrier. Brain Res 381: 131-137.
- Beck DW, Vinters HV, Hart MN, Cancilla PA (1984) Glial cells influence polarity of the blood-brain barrier. J Neuropathol Exp Neurol 43: 219-224.
- Tontsch U, Bauer HC (1991) Glial cells and neurons induce blood-brain barrier related enzymes in cultured cerebral endothelial cells. Brain Res 539: 247-253.
- Cho H, Seo JH, Wong KHK, Terasaki Y, Park J, et al. (2015) Three-dimensional blood-brain barrier model for in vitro studies of neurovascular pathology. Sci Rep 5: 15222.
- Ishibashi T, Dakin KA, Stevens B, Lee PR, Kozlov SV, et al. (2006) Astrocytes promote myelination in response to electrical impulses. Neuron 49: 823-832.
- Kilic O, Pamies D, Lavell E, Schiapparelli P, Feng Y, et al. (2016) Brain-on-a-chip model enables analysis of human neuronal differentiation and chemotaxis. Lab Chip 16: 4152-4162.
- Brightman MW, Reese TS, Vick NA, Bigner DD (1971) A mechanism underlying the lack of a blood-brain barrier to peroxidase in virally induced brain tumors. J Neuropathol Exp Neurol 30: 139-140.
- Brightman MW, Klatzo I, Olsson Y, Reese TS (1970) The blood-brain barrier to proteins under normal and pathological conditions. J Neurol Sci 10: 215-239.
- Reese TS, Karnovsky MJ (1967) Fine structural localization of a blood-brain barrier to exogenous peroxidase. J Cell Biol 34: 207-217.
- Seo JH, Maki T, Maeda M, Miyamoto N, Liang AC, et al. (2014) Oligodendrocyte precursor cells support blood-brain barrier integrity via TGF-β signaling. PLoS One 9: e103174.
- Appelt-Menzel A, Cubukova A, Günther K, Edenhofer F, Piontek J, et al. (2017) Establishment of a human blood-brain barrier co-culture model mimicking the neurovascular unit using induced pluri- and multipotent stem cells. Stem Cell Reports 8: 894-906.
- Arthur FE, Shivers RR, Bowman PD (1987) Astrocyte-mediated induction of tight junctions in brain capillary endothelium: An efficient in vitro model. Brain Res 433: 155-159.
- Cancilla PA, DeBault LE (1983) Neutral amino acid transport properties of cerebral endothelial cells in vitro. J Neuropathol Exp Neurol 42: 191-199.
- Muoio V, Persson PB, Sendeski MM (2014) The neurovascular unit-concept review. Acta physiologica 210: 790-798.
- Girouard H, Iadecola C (2006) Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol (1985) 100: 328-335.
- Buerk DG, Ances BM, Greenberg JH, Detre JA (2003) Temporal dynamics of brain tissue nitric oxide during functional forepaw stimulation in rats. Neuroimage 18: 1-9.
- Lindauer U, Megow D, Matsuda H, Dirnagl U (1999) Nitric oxide: A modulator, but not a mediator, of neurovascular coupling in rat somatosensory cortex. Am J Physiol277: H799-H811.
- Park L, Anrather J, Forster C, Kazama K, Carlson GA, et al. (2004) Aβ-induced vascular oxidative stress and attenuation of functional hyperemia in mouse somatosensory cortex. J Cereb Blood Flow Metab 24: 334-342.
- Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ (2010) Structure and function of the blood-brain barrier. Neurobiol Dis 37: 13-25.
- Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, et al. (2010) Pericytes regulate the blood-brain barrier. J Nature 468: 557-561.
- Ballabh P, Braun A, Nedergaard M (2004) The blood-brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol Dis 16: 1-13.
- Ben-Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, et al. (2014) Mfsd2a is critical for the formation and function of the blood-brain barrier. J Nature 509: 507.
- Liebner S, Dijkhuizen RM, Reiss Y, Plate KH, Agalliu D, et al. (2018) Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathologica 135: 311-336.
- Vandenhaute E, Dehouck L, Boucau MC, Sevin E, Uzbekov R, et al. (2011) Modelling the neurovascular unit and the blood-brain barrier with the unique function of pericytes. Curr Neurovasc Res 8: 258-269.
- Yamazaki Y, Kanekiyo T (2017) Blood-brain barrier dysfunction and the pathogenesis of Alzheimer’s disease. Int J Mol Sci 18: 1965.
- Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, et al. (1998) Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 91: 3527-3561.
- Pittman RN (2011) Regulation of tissue oxygenation. in Colloquium series on integrated systems physiology: From molecule to function. 2011. Morgan & Claypool Life Sciences.
- Schaeffer M, Hodson DJ, Lafont C, Mollard P (2011) Endocrine cells and blood vessels work in tandem to generate hormone pulses. J Mol Endocrinol47: R59-R66.
- Günzel D, Yu AS (2013) Claudins and the modulation of tight junction permeability. Physiol Rev 93: 525-569.
- Liebner S, Corada M, Bangsow T, Babbage J, Taddei A, et al. (2008) Wnt/β-catenin signaling controls development of the blood-brain barrier. J Cell Biol 183: 409-417.
- Liebner S, Fischmann A, Rascher G, Duffner F, Grote EH, et al. (2000) Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropatho 100: 323-331.
- Morita K, Sasaki H, Furuse M, Tsukita S (1999) Endothelial claudin: Claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol 147: 185-194.
- Tsukita S, Furuse M (1999) Occludin and claudins in tight-junction strands: leading or supporting players? Trends Cell Biol 9: 268-273.
- Wolburg H, Wolburg-Buchholz K, Kraus J, Rascher-Eggstein G, Liebner S, et al. (2003) Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathol 105: 586-592.
- Taddei A, Giampietro C, Conti A, Orsenigo F, Breviario F, et al. (2008) Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol 10: 923.
- McCarthy KM, Skare IB, Stankewich MC, Furuse M, Tsukita S, et al. (1996) Occludin is a functional component of the tight junction. J Cell Sci 109: 2287-2298.
- Wong V, Gumbiner BM (1997) A synthetic peptide corresponding to the extracellular domain of occludin perturbs the tight junction permeability barrier. J Cell Biol 136: 399-409.
- Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, et al. (2000) Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell 11: 4131-4142.
- Tornavaca O, Chia M, Dufton N, Almagro LO, Conway DE, et al. (2015) ZO-1 controls endothelial adherens junctions, cell–cell tension, angiogenesis, and barrier formation. J Cell Biol 208: 821-838.
- Furuse M (2014) Molecular organization of tricellular tight junctions. Yakugaku Zasshi 134: 615-621.
- Ikenouchi J, Furuse M, Furuse K, Sasaki H, Tsukita S, et al. (2005) Tricellulin constitutes a novel barrier at tricellular contacts of epithelial cells. J Cell Biol 171: 939-945.
- Masuda S, Oda Y, Sasaki H, Ikenouchi J, Higashi T, et al. (2011) LSR defines cell corners for tricellular tight junction formation in epithelial cells. J Cell Sci 124: 548-555.
- Vestweber D (2008) VE-cadherin: The major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arterioscler Thromb Vasc Biol 28: 223-232.
- Hawkins BT, Davis TP (2005) The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev 57: 173-185.
- Gavard J, Gutkind JS (2008) VE-cadherin and claudin-5: It takes two to tango. Nat Cell Biol 10: 808-883.
- Oldendorf WH, Cornford ME, Brown WJ (1977) The large apparent work capability of the blood?brain barrier: A study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann Neurol 1: 409-417.
- Boado RJ, Pardridge WM (1993) Glucose deprivation causes posttranscriptional enhancement of brain capillary endothelial glucose transporter gene expression via GLUT1 mRNA stabilization. J Neurochem 60: 2290-2296.
- Cardoso FL, Brites D, Brito MA (2010) Looking at the blood-brain barrier: Molecular anatomy and possible investigation approaches. Brain Res Rev 64: 328-363.
- Zlokovic BV (2008) The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57: 178-201.
- Begley DJ (2004) ABC transporters and the blood-brain barrier. Curr Pharm Des 10: 1295-1312.
- Dauchy S, Dutheil F, Weaver RJ, Chassoux F, Daumas-Duport C, et al. (2008) ABC transporters, cytochromes P450 and their main transcription factors: Expression at the human blood-brain barrier. J Neurochem107: 1518-1528.
- Miller DS (2010) Regulation of P-glycoprotein and other ABC drug transporters at the blood-brain barrier. Trends Pharmacol Sci 31: 246-254.
- Huber JD, Egleton RD, Davis TP (2001) Molecular physiology and pathophysiology of tight junctions in the blood-brain barrier. Trends Neurosci 24: 719-725.
- Preston JE, Joan Abbott N, Begley DJ (2014) Transcytosis of macromolecules at the blood-brain barrier.Adv Pharmacol 71: 147-163.
- Nguyen LN, Ma D, Shui G, Wong P, Cazenave-Gassiot A, et al. (2014) Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 509:503.
- Quek DQ, Nguyen LN, Fan H, Silver DL (2016) Structural insights into the transport mechanism of the human sodium-dependent lysophosphatidylcholine transporter Mfsd2a. J Biol Chem 291: 9383-9394.
- Alvarez JI, Saint-Laurent O, Godschalk A, Terouz S, Briels C, et al. (2015) Focal disturbances in the blood-brain barrier are associated with formation of neuroinflammatory lesions. Neurobiol Dis 74: 14-24.
- Zhao YL, Song JN, Zhang M (2014) Role of caveolin-1 in the biology of the blood-brain barrier. Rev Neurosci 25: 247-254.
- Kronstein R, Seebach J, Grossklaus S, Minten C, Engelhardt B, et al. (2011) Caveolin-1 opens endothelial cell junctions by targeting catenins. Cardiovasc Res 93: 130-140.
- Stamatovic SM, Keep RF, Wang MM, Jankovic I, Andjelkovic AV (2009) Caveolae?mediated internalization of occludin and claudin?5 during CCL2?induced tight junction remodeling in brain endothelial cells. J Biol Chem 284: 19053-19066.
- Betz AL, Firth JA, Goldstein GW (1980) Polarity of the blood-brain barrier: Distribution of enzymes between the luminal and antiluminal membranes of brain capillary endothelial cells. Brain Res 192: 17-28.
- Meyer J, Mischeck U, Veyhl M, Henzel K, Galla HJ (1990) Blood-brain barrier characteristic enzymatic properties in cultured brain capillary endothelial cells. Brain Res 514: 305-309.
- Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD (2006) Blood-brain barrier: Structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol 1: 223-236.
- Sá-Pereira I, Brites D, Brito MA (2012) Neurovascular unit: A focus on pericytes. Mol Neurobiol 45: 327-347.
- Elfont RM, Sundaresan PR, Sladek CD (1989) Adrenergic receptors on cerebral microvessels: Pericyte contribution. Am J Physiol 256: R224-R230.
- Healy DP, Wilk S (1993) Localization of immunoreactive glutamyl aminopeptidase in rat brain. II. Distribution and correlation with angiotensin II. Brain Res 606: 295-303.
- Benagiano V, Virgintino D, Maiorano E, Rizzi A, Palombo S, et al. (1996) VIP-like immunoreactivity within neurons and perivascular neuronal processes of the human cerebral cortex. Eur J Histochem 40: 53-56.
- Dehouck MP, Vigne P, Torpier G, Breittmayer JP, Cecchelli R, et al. (1997) Endothelin-1 as a mediator of endothelial cell-pericyte interactions in bovine brain capillaries. J Cereb Blood Flow Metab17: 464-469.
- van Zwieten EJ, Ravid R, Swaab DF, Van de Woude T (1988) Immunocytochemically stained vasopressin binding sites on blood vessels in the rat brain. Brain Res 474: 369-373.
- Ramsauer M, Krause D, Dermietzel R (2002) Angiogenesis of the blood-brain barrier in vitro and the function of cerebral pericytes. FASEB J 16: 1274-1276.
- Sweeney MD, Ayyadurai S, Zlokovic BV (2006) Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat Neurosci 19: 771-783.
- Gautam JX, Zhang X, Yao Y (2016) The role of pericytic laminin in blood brain barrier integrity maintenance. Sci Rep 6: 36450.
- Morris AW, Carare RO, Schreiber S, Hawkes CA (2014) The cerebrovascular basement membrane: Role in the clearance of β-amyloid and cerebral amyloid angiopathy. Front Aging Neurosci 6: 251.
- Thomsen MS, Routhe LJ, Moos T (2017) The vascular basement membrane in the healthy and pathological brain. J Cereb Blood Flow Metab 37: 3300-3317.
- Yousif LF, Di Russo J, Sorokin L (2013) Laminin isoforms in endothelial and perivascular basement membranes. Cell Adh Migr 7: 101-110.
- Baeten KM, Akassoglou K (2011) Extracellular matrix and matrix receptors in blood-brain barrier formation and stroke. Dev Neurobiol 71: 1018-1039.
- Owens T, Bechmann I, Engelhardt B (2008) Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol 67: 1113-1121.
- Sixt M, Engelhardt B, Pausch F, Hallmann R, Wendler O, et al. (2001) Endothelial cell laminin isoforms, laminins 8 and 10, play decisive roles in T cell recruitment across the blood-brain barrier in experimental autoimmune encephalomyelitis. J Cell Biol 153: 933-946.
- Zhang ET, Inman CB, Weller RO (1990) Interrelationships of the pia mater and the perivascular (Virchow-Robin) spaces in the human cerebrum. J Anat 170: 111-123.
- Sorokin L (2010) The impact of the extracellular matrix on inflammation. Nat Rev Immunol 10: 712-723.
- Savettieri G, Di Liegro I, Catania C, Licata L, Pitarresi GL, et al. (2000) Neurons and ECM regulate occludin localization in brain endothelial cells. Neuroreport 11: 1081-1084.
- Tilling T, Korte D, Hoheisel D, Galla HJ (1998) Basement membrane proteins influence brain capillary endothelial barrier function in vitro. J Neurochem 71: 1151-1157.
- Brown JA, Codreanu SG, Shi M, Sherrod DS, Markov DA, et al. (2016) Metabolic consequences of inflammatory disruption of the blood-brain barrier in an organ-on-chip model of the human neurovascular unit. J Neuroinflammation 13: 306.
- Ruoslahti E (1996) Brain extracellular matrix. Glycobiology 6: 489-492.
- Engelhardt B, Liebner S (2014) Novel insights into the development and maintenance of the blood-brain barrier. Cell Tissue Res 355: 687-699.
- Abbott NJ, Dolman DE, Drndarski S, Fredriksson SM (2012) An improved in vitro blood-brain barrier model: Rat brain endothelial cells co-cultured with astrocytes, in astrocytes. Methods Mol Biol 814: 415-430.
- Abbott NJ, Rönnbäck L, Hansson E (2006) Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 7: 41-43.
- Gaillard PJ, Voorwinden LH, Nielsen JL, Ivanov A, Atsumi R, et al. (2001) Establishment and functional characterization of an in vitro model of the blood-brain barrier, comprising a co-culture of brain capillary endothelial cells and astrocytes. Eur J Pharm Sci 12: 215-222.
- Nakagawa S, Deli MA, Kawaguchi H, Shimizudani T, Shimono T, et al. (2009) A new blood-brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochem Int 54: 253-263.
- Zhou Y, Wang Y, Tischfield M, Williams J, Smallwood PM, et al. (2014) Canonical WNT signaling components in vascular development and barrier formation. J Clin Invest 124: 3825-3846.
- Alvarez JI, Dodelet-Devillers A, Kebir H, Ifergan I, Fabre PJ, et al. (2011) The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 334: 1727-1731.
- Gurnik S, Devraj K, Macas J, Yamaji M, Starke J, et al. (2016) Angiopoietin-2-induced blood-brain barrier compromise and increased stroke size are rescued by VE-PTP-dependent restoration of Tie2 signaling. Acta Neuropathol 131: 753-773.
- Sandoval KE, Witt KA (2008) Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol Dis 32: 200-219.
- Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308: 1314-1318.
- Kreutzberg GW (1996) Microglia: A sensor for pathological events in the CNS. Trends Neurosci 19: 312-318.
- Schroeter M, Jander S, Witte OW, Stoll G (1999) Heterogeneity of the microglial response in photochemically induced focal ischemia of the rat cerebral cortex. Neuroscience 89: 1367-1377.
- Streit WJ, Mrak RE, Griffin WST (2004) Microglia and neuroinflammation: A pathological perspective. J Neuroinflammation 1: 14.
- Streit WJ (2002) Microglia as neuroprotective, immunocompetent cells of the CNS. Glia 40: 133-139.
- Nolte C, Möller T, Walter T, Kettenmann H (1996) Complement 5a controls motility of murine microglial cells in vitro via activation of an inhibitory G-protein and the rearrangement of the actin cytoskeleton. Neuroscience 73: 1091-1107.
- Stence N, Waite M, Dailey ME (2001) Dynamics of microglial activation: A confocal time?lapse analysis in hippocampal slices. Glia 33: 256-266.
- Garden GA, Möller T (2006) Microglia biology in health and disease. J Neuroimmune Pharmacol 1: 127-137.
- Kim SU, de Vellis J (2005) Microglia in health and disease. J Neurosci Res 81: 302-313.
- Ransohoff RM, Khoury JEL (2016) Microglia in health and disease. Cold Spring Harb Perspect Biol 8: a020560.
- Csuka E, Morganti-Kossmann MC, Lenzlinger PM, Joller H, Trentz O, et al. (1999) IL-10 levels in cerebrospinal fluid and serum of patients with severe traumatic brain injury: relationship to IL-6, TNF-α, TGF-β1 and blood–brain barrier function. J Neuroimmunol 101: 211-221.
- Nishioku T, Matsumoto J, Dohgu S, Sumi N, Miyao K, et al. (2010) Tumor necrosis factor-α mediates the blood–brain barrier dysfunction induced by activated microglia in mouse brain microvascular endothelial cells. J Pharmacol Sci 112: 251-254.
- Baumann N, Pham-Dinh D (2001) Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev 81: 871-927.
- Rhodes KE, Raivich G, Fawcett JW (2006) The injury response of oligodendrocyte precursor cells is induced by platelets, macrophages and inflammation-associated cytokines. Neuroscience 140: 87-100.
- Banerjee S, Bhat MA (2007) Neuron-glial interactions in blood-brain barrier formation. Annu Rev Neurosci 30: 235-258.
- Lippmann ES, Weidenfeller C, Svendsen CN, Shusta EV (2011) Blood-brain barrier modeling with co?cultured neural progenitor cell?derived astrocytes and neurons. J Neurochem 119: 507-520.
- Minami MJ (2011) Neuro-glio-vascular interaction in ischemic brains. Journal of the Pharmaceutical Society of Japan 131: 539-544.
- Stolp HB, Dziegielewska KM (2009) Role of developmental inflammation and blood-brain barrier dysfunction in neurodevelopmental and neurodegenerative diseases. Neuropathol Appl Neurobiol 35: 132-146.
- Zlokovic BV (2010) Neurodegeneration and the neurovascular unit. Nat Med 16: 1370-1371.
- Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV (2016) Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer's disease. Biochim Biophys Acta 1862: 887-900.
- Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140: 918-934.
- Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, et al. (2009) The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 132: 1175-1189.
- Block ML, Hong JS (2005) Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog Neurobiol 76: 77-98.
- McGeer PL, McGeer EG (2004) Inflammation and neurodegeneration in Parkinson's disease. Parkinsonism Relat Disord 10: S3-S7.
- Garbuzova-Davis S, Saporta S, Sanberg PR (2008) Implications of blood-brain barrier disruption in ALS. Amyotroph Lateral Scler 9: 375-376.
- Garbuzova-Davis S, Haller E, Saporta S, Kolomey I, Nicosia SV, et al. (2007) Ultrastructure of blood-brain barrier and blood-spinal cord barrier in SOD1 mice modeling ALS. Brain Res 1157: 126-137.
- Nicaise C, Soyfoo MS, Delporte C, Pochet R (2010) Aquaporin-4 as a potential marker of BBB disruption in ALS models. Amyotroph Lateral Scler 11: 253-254.
- Lin CY, Hsu YH, Lin MH, Yang TH, Chen HM, et al. (2013) Neurovascular abnormalities in humans and mice with Huntington's disease. Exp Neurol 250: 20-30.
- Lim RG, Quan C, Reyes-Ortiz AM, Lutz SE, Kedaigle AJ, et al. (2017) Huntington’s disease iPSC-derived brain microvascular endothelial cells reveal WNT-mediated angiogenic and blood-brain barrier deficits. Cell Rep 19: 1365-1377.
- Di Pardo A, Amico E, Scalabrì F, Pepe G, Castaldo S, et al. (2017) Impairment of blood-brain barrier is an early event in R6/2 mouse model of Huntington Disease. Sci Rep 7: 41316.
- Avison MJ, Nath A, Greene-Avison R, Schmitt FA, Greenberg RN, et al. (2004) Neuroimaging correlates of HIV-associated BBB compromise. J Neuroimmunol 157: 140-146.
- Kanmogne GD, Schall K, Leibhart J, Knipe B, Gendelman HE, et al. (2007) HIV-1 gp120 compromises blood–brain barrier integrity and enhance monocyte migration across blood-brain barrier: Implication for viral neuropathogenesis. J Cereb Blood Flow Metab 27: 123-134.
- Ricardo-Dukelow M, Kadiu I, Rozek W, Schlautman J, Persidsky Y, et al. (2007) HIV-1 infected monocyte-derived macrophages affect the human brain microvascular endothelial cell proteome: New insights into blood-brain barrier dysfunction for HIV-1-associated dementia. J Neuroimmunol 185: 37-46.
- Moses AV, Nelson JA (1994) HIV infection of human brain capillary endothelial cell-Implications for AIDS dementia. Adv Neuroimmunol 4: 239-247.
- Schneck MK, Reisberg B, Ferris SH (1982) An overview of current concepts of Alzheimer's disease. Am J Psychiatry 139: 165-173.
- Montagne A, Zhao Z, Zlokovic BV (2017) Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J Exp Med 214: 3151-3169.
- Toledo JB, Arnold SE, Raible K, Brettschneider J, Xie SX, et al. (2013) Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain 136: 2697-2706.
- Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Pérez JM, Evans AC, et al. (2016) Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun 7: 11934.
- Faraco G, Park L, Zhou P, Luo W, Paul SM, et al. (2016) Hypertension enhances A β-induced neurovascular dysfunction, promotes β-secretase activity, and leads to amyloidogenic processing of APP. J Cereb Blood Flow Metab 36: 241-252.
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, et al. (1995) Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 373: 523-527.
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, et al. (1998) Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 4: 97-100.
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, et al. (2003) Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron 39: 409-421.
- Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, et al. (2000) Hypercholesterolemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model. Neurobiol Dis 7: 321-331.
- Biron KE, Dickstein DL, Gopaul R, Jefferies (2011)Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer's disease. PLoS One 6: 23789.
- Kumar-Singh S, Pirici D, McGowan E, Serneels S, Ceuterick C, et al. (2005) Dense-core plaques in Tg2576 and PSAPP mouse models of Alzheimer's disease are centered on vessel walls. Am J Pathol 167: 527-543.
- Park L, Zhou J, Zhou P, Pistick R, El Jamal S, et al. (2013) Innate immunity receptor CD36 promotes cerebral amyloid angiopathy. Proc Natl Acad Sci U S A 110: 3089-3094.
- Paul J, Strickland S, Melchor JP (2007) Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer's disease. J Exp Med 204: 1999-2008.
- Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, et al. (2013) Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat Commun 4: 2932.
- Ujiie M, Dickstein DL, Carlow DA, Jefferies WA (2003) Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation 10: 463-470.
- Deane R, Wu Z, Sagare A, Davis J, Du Yan S, et al. (2004) LRP/amyloid β-peptide interaction mediates differential brain efflux of Aβ Neuron 43: 333-344.
- Kook SY, Hong HS, Moon M, Ha CM, Chang S, et al. (2012) Aβ1-42-RAGE interaction disrupts tight junctions of the blood–brain barrier via Ca2+-calcineurin signaling. J Neurosci 32: 8845-8854.
- Niwa K1, Kazama K, Younkin L, Younkin SG, Carlson GA, et al. (2002) Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. Am J Physiol Heart Circ Physiol283: 315-323.
- Winkler EA, Nishida Y, Sagare AP, Rege SV, Bell RD, et al. (2015) GLUT1 reductions exacerbate Alzheimer's disease vasculo-neuronal dysfunction and degeneration. Nat Neurosci 18: 521-530.
- Gama Sosa MA, Gasperi RD, Rocher AB, Wang AC, Janssen WG, et al. (2010) Age-related vascular pathology in transgenic mice expressing presenilin 1-associated familial Alzheimer's disease mutations. Am J Pathol 176: 353-368.
- Boekhoorn K, Terwel D, Biemans B, Borghgraef P, Wiegert O, et al. (2006) Improved long-term potentiation and memory in young tau-P301L transgenic mice before onset of hyperphosphorylation and tauopathy. J Neurosci 26: 3514-3523.
- Fullerton SM, Shirman GA, Strittmatter WJ, Matthew WD (2001) Impairment of the blood-nerve and blood-brain barriers in apolipoprotein E knockout mice. Exp Neurol 169: 13-22.
- Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, et al. (2015) Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85: 296-302.
- van de Haar HJ, Burgmans S, Jansen JF, van Osch MJ, van Buchem MA, et al. (2016) Blood-brain barrier leakage in patients with early Alzheimer disease. Radiology 281: 527-535.
- Zhang CE, Wong SM, van de Haar HJ, Staals J, Jansen JF, et al. (2017) Blood-brain barrier leakage is more widespread in patients with cerebral small vessel disease. Neurology 88: 426-432.
- Cabezas R, Avila M, Gonzalez J, El-Bachá RS, Báez E, et al. (2014) Astrocytic modulation of blood brain barrier: perspectives on Parkinson’s disease. Front Cell Neurosci 8: 211.
- Halliday GM, Stevens CH (2011) Glia: initiators and progressors of pathology in Parkinson's disease. Mov Disord 26: 6-17.
- Nutt JG, Wooten GF (2005) Diagnosis and Initial Management of Parkinson's Disease. New England Journal of Medicine 353: 1021-1027.
- Fernandez HH (2012) Updates in the medical management of Parkinson disease. Cleve Clin J Med 79: 28-35.
- Singer C (2012) Managing the patient with newly diagnosed Parkinson disease. Cleve Clin J Med 79: 3-7.
- Jenner P (2003) Oxidative stress in Parkinson's disease. Ann Neurol 53: 26-38.
- Vives-Bauza C, Przedborski S (2011) Mitophagy: the latest problem for Parkinson's disease. Trends Mol Med 17: 158-165.
- Braak H, de Vos RA, Bohl J, Del Tredici K (2006) Gastric alpha-synuclein immunoreactive inclusions in Meissner's and Auerbach's plexuses in cases staged for Parkinson's disease-related brain pathology. Neurosci Lett 396: 67-72.
- Jangula A, Murphy EJ (2013) Lipopolysaccharide-induced blood brain barrier permeability is enhanced by alpha-synuclein expression. Neurosci Lett 551: 23-27.
- Haussermann 1, Kuhn W, Przuntek H, Muller T (2001) Integrity of the blood–cerebrospinal fluid barrier in early Parkinson's disease. Neurosci Lett 300: 182-184.
- Kurkowska-Jastrzebska I, Wro?ska A, Kohutnicka M, Cz?onkowski A, Cz?onkowska A (1999) The inflammatory reaction following 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine intoxication in mouse. Exp Neurol 156: 50-61.
- Hirano T, Sasaki M, Mori E, Minematsu K, Nakagawara J, et al. (2010) Residual vessel length on magnetic resonance angiography identifies poor responders to alteplase in acute middle cerebral artery occlusion patients: exploratory analysis of the Japan Alteplase Clinical Trial II. Stroke 41: 2828-2833.
- Kortekaas R, Leenders KL, van Oostrom JC, Vaalburg W, Bart J, et al. (2005) Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol 57: 176-179.
- Lee H, Pienaar IS (2014) Disruption of the blood-brain barrier in Parkinson’s disease: curse or route to a cure. Front Biosci 19: 272-280.
- Ohlin KE, Francardo V, Lindgren HS, Sillivan SE, O'Sullivan SS, et al. (2011) Vascular endothelial growth factor is upregulated by L-dopa in the parkinsonian brain: implications for the development of dyskinesia. Brain 134: 2339-2357.
- Bartels AL, Willemsen AT, Kortekaas R, de Jong BM, de Vries R, et al. (2008) Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J Neural Transm (Vienna) 115: 1001-1009.
- Schinkel AH, Smit JJ, van Tellingen O, Beijnen JH, Wagenaar E, et al. (1994) Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell 77: 491-502.
- Popescu BO, Toescu EC, Popescu LM, Bajenaru O, Muresanu DF, et al. (2009) Blood-brain barrier alterations in ageing and dementia. J Neurol Sci 283: 99-106.
- Enciu AM, Popescu BO (2013) Is there a causal link between inflammation and dementia? Biomed Res Int316495.
- Fukui K, Onodera K, Shinkai T, Suzuki S, Urano S (2001) Impairment of learning and memory in rats caused by oxidative stress and aging, and changes in antioxidative defense systems. Ann N Y Acad Sci 928: 168-175.
- Calabresi PA (2004) Diagnosis and management of multiple sclerosis. Am Fam Physician 70: 1935-1944.
- Hauser SL Goodin DS (1994) Multiple sclerosis and other demyelinating diseases. Harrison's principles of internal medicine: 2287.
- Spencer JI, Bell JS, DeLuca GC (2018) Vascular pathology in multiple sclerosis: reframing pathogenesis around the blood-brain barrier. J Neurol Neurosurg Psychiatry 89: 42-52.
- Zlokovic BV (2008) The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron, 57: 178-201.
- Ransohoff RM (2007) Natalizumab for multiple sclerosis. N Engl J Med 356: 2622-2629.
- Engelhardt B, Ransohoff RM (2005) The ins and outs of T-lymphocyte trafficking to the CNS: Anatomical sites and molecular mechanisms. Trends Immunol 26: 485-495.
- Krumbholz M, Theil D, Cepok S, Hemmer B, Kivisäkk P, et al. (2005) Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain 129: 200-211.
- Kivisäkk P, Mahad DJ, Callahan MK, Trebst C, Tucky B, et al. (2003) Human cerebrospinal fluid central memory CD4+ T cells: Evidence for trafficking through choroid plexus and meninges via P-selectin. Proc Natl Acad Sci U S A 100: 8389-8394.
- Ransohoff RM, Kivisäkk P, Kidd G (2003) Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol 3: 569-581.
- Man S, Tucky B, Bagheri N, Li X, Kochar R, et al. (2009) α4 Integrin/FN-CS1 mediated leukocyte adhesion to brain microvascular endothelial cells under flow conditions. J Neuroimmunol 210: 92-99.
- Mahad DJ, Ransohoff RM (2003) The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Semin Immunol 15: 23-32.
- Nzou G, Wicks RT, Wicks EE, Seale SE, Sane CH, et al. (2018) Human cortex spheroid with a functional blood brain barrier for high-throughput neurotoxicity screening and disease modeling. Sci Rep 8: 7413.
- Liow JS, Lu S, McCarron JA, Hong J, Musachio JL, et al. (2007) Effect of a P?glycoprotein inhibitor, cyclosporin A, on the disposition in rodent brain and blood of the 5?HT1A receptor radioligand,[11C](R)?(––)?RWAY. Synapse 61: 96-105.
- Syvänen S, Lindhe O, Palner M, Kornum BR, Rahman O, et al. (2009) Species differences in blood-brain barrier transport of three positron emission tomography radioligands with emphasis on P-glycoprotein transport. Drug Metab Dispos 37: 635-643.
- Yasuno F, Zoghbi SS, McCarron JA, Hong J, Ichise M, et al. (2006) Quantification of serotonin 5?HT1A receptors in monkey brain with [11C](R)?(−)?RWAY. Synapse 60: 510-520.
- Zhang Xy, Yasuno F, Zoghbi SS, Liow JS, Hong J, et al. (2007) Quantification of serotonin 5?HT1A receptors in humans with [11C](R)?(−)?RWAY: Radiometabolite (s) likely confound brain measurements. Synapse 61: 469-477.
- Hood L, Friend SH (2011) Predictive, personalized, preventive, participatory (P4) cancer medicine. Nat Rev Clin Oncol 8: 184-187.
- Chin L, Andersen JN, Futreal PA (2011) Cancer genomics: From discovery science to personalized medicine. Nat Med 17: 297-303.
- Verma M (2012) Personalized medicine and cancerJ Pers Med 2: 1-14.
- Ross JS, Slodkowska EA, Symmans WF, Pusztai L, Ravdin PM, et al. (2009) The HER-2 receptor and breast cancer: Ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist 14: 320-368.
- Mattson MP, Duan W, Pedersen WA, Culmsee C (2001) Neurodegenerative disorders and ischemic brain diseases. Apoptosis 6: 69-81.
- Frank SA (2010) Evolution in health and medicine Sackler colloquium: Somatic evolutionary genomics: mutations during development cause highly variable genetic mosaicism with risk of cancer and neurodegeneration. Proc Natl Acad Sci U S A 107: 1725-1730.
- Campbell A (2004) Inflammation, neurodegenerative diseases, and environmental exposures. Ann N Y Acad Sci 1035: 117-132.
Citation: Nzou G, Seeds MC, Wicks RT, Atala AJ (2019) Fundamental Neurovascular Components for the Development of Complex and Dynamic in Vitro Brain Equivalent Models. J Alzheimers Neurodegener Dis 5: 021.
Copyright: © 2019 Goodwell Nzou, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Journal Highlights
© 2024, Copyrights Herald Scholarly Open Access. All Rights Reserved!