Cancer Stem Cells: Understanding their Dynamics
*Corresponding Author(s):
Shailja ChatterjeeDepartment Of Oral And Maxillofacial Pathology, MM College Of Dental Sciences And Research, Maharishi Markandeshwar University, Mullana, Haryana, India
Tel:+91 9412743252,
Email:shailjachatterjee@gmail.com
Abstract
Cancer stem cells are specialized cells which have different genetic constitution than the stem cells residing in normal tissues. This article discusses various hierarchial models that conceptualize cancer stem cell development and their role in tumor development. These cells are important in terms of therapeutic intervention as these remain as residual disease in treated sites, which can reappear in form of recurrence and secondaries. Understanding the stem cell niche is an important aspect of regulatory mechanisms operating in the immediate microenvironment. This review covers the molecular mechanisms and models regulating Cancer stem cell development, tumor initiation, progression and metastasis providing the reader an insight into understanding these entities.
Keywords
INTRODUCTION
The term “stem cell” was first used by Russian Researcher, Alexander A Maximow as early as 1909 [1]. The normal adult stem cells and cancer stem cells share important properties like-self-renewal capacity, differentiation ability, active telomerase expression, activation of antiapoptotic pathway, increased membrane transporter activity and ability to migrate [2,3].
Cancer stem cells are small primitive cell population, which are quiescent, resistant to therapy, capable of self-renewal and play an important role in initiating cancer. Two models that can explain the biological behaviour of these cells are: a) Stochastic model, and b) Hierarchy model. According to the stochastic model, tumors are biologically homogenous with cancer stem cell behaviour being governed by intrinsic and/or extrinsic behaviour. Whereas, the hierarchy model suggests that the cancer stem cell are biologically unique. The ‘cancer stem cell hypothesis’ explains residual disease, disease relapse, disease progression thus, highlighting the requirement of targeting these cells for an effective cure. Normal epithelial cells undergo apoptosis after their detachment from the extracellular matrix. This collective process in termed as the ‘anoikis’. Cancer cells’ subset at the invasive front in breast carcinoma displays an altered genetic expression of the epithelial and mesenchymal components. This process in termed as “Epithelial-Mesenchymal transition”. This transformation confers properties of invasiveness, resistance to chemotherapy, recurrence and tumorigenesis. Thus, anoikis resistance can be attributed to epithelial-mesenchymal transformation. Molecular mechanisms responsible for this transformation factor activity and pro-survival gene activation are the results of epithelial-mesenchymal transformation. High CD44 expression has been related to metastasis and disease progression in various cancer types [4].
Cancer evolution is a step-wise, nonlinear cellular differentiation process. Genetically abnormal clonal cancer cell populations might maintain a quiescent state for a long period of time. Neoplastic cell clones contain aberrations required for development of malignancy while cancer stem cells are restricted towards generation of sub-clones which aid in maintenance of cancerous clones. Due to self-renewal property of stem cells, unrepaired genomic damage is inherent in the amplified progeny. The host immune response is effective in eliminating these deranged cells. However, with increasing age and diminished immunocompetence, this elimination process remains incomplete leading to accumulation of pro-angiogenic accumulative changes. During early phase of neoplastic clone development, the minimally acquired mutations have very little effect on morphologic, immunological and transcriptional features [5].
The cancer stem cells are characterized by three features: 1) They exhibit slight resemblance to the tissue of origin; 2) Permanency of malignant cell population; and 3) Multiplicity of genetic events which accumulate this gives rise to a malignant cell population [6].
MODELS OF CANCER INITIATION AND PROGRESSION
Nowell and Vogelstein postulated the ‘Carcinogenesis Model’, which describes tumor formation as a result of sequential accumulation of genetic mutations in oncogenes and tumor suppressor genes [7,8]. According to this model, tumors are believed to comprise of a heterogenous cell population that acquires new mutations and results in formation of a mutated clonal population. The clonal selection theory proposed by Nowell views malignancy as a clonally derived cell population with mutations that result in proliferation of new mutated clones [6].
There are two mechanisms of cancer initiation and progression.
According to the first mechanism
Cancer stem cell hypothesis: According to this hypothesis, the mutations giving rise to cancer occur in normal adult stem cells. These cells are self-renewing precursor cells responsible for cancer stem cell generation and are genetically identical to cancer stem cells.
According to the second mechanism
Clonal evolution model: Any normal cell is a target for transformation. The cancer cell acquires additional mutations, which give rise to a line of cancerous cells. This process in repeated infinitely till a tumor mass is detectable. Therapeutic implication as per cancer stem cell hypothesis states that elimination of a cancer stem cell results in the cessation of tumor growth, thus, curing the disease [9].
Hematological malignant stem cells and their markers
The hematological stem cells were first identified by Weismann et al. These stem cell populations give rise to a more committed progenitor cell population that result in various cell components like- B cells, T cells and macrophages [10]. Leukemias contain a diversity of cells but are monoclonal in origin [7]. These cells reside in a CD34+/Lin- subset of malignant clones [9,10].
Membranous markers for identification of cancer stem cell population include CD133, CD44 and CD24. Metastatic cancer stem cell represents a dominant clone in a tumor population. A cancer stem cell on seeding reaches a distant site where the microenvironment stimulates the cancer stem cell growth with few added genetic mutations (Paget’s seed and soil theory). The cell adhesion receptor, CD44 is a lymphocyte homing receptor for the ligand, ‘hyaluronan’. Multiple CD44 isoforms arise from differential splicing. These isoforms are expressed in specific tissues or various cell types. The CD44 gene promoter is positively regulated by p63 protein and Wnt signaling. P53 causes promoter repression by preventing p63 recruitment. CD44 upregulation correlates with angiogenesis in breast carcinoma. CD44-hyaluronic acid interaction stimulates cell migration and invasion through Ezrin/redixin/Moesin protein, ankyrin-G and rhoA. The isoforms CD44S protects the epithelia-mesenchymal transformed tumor cells against anoikis. CD44S interacts with its ligand, hyaluronan, thus enabling metastasis [9].
CD44 is a homing cell adhesion molecule which is biochemically a cell surface glycoprotein expressed on lymphocytes, monocytes and granulocytes. It has been recognized as a cancer stem cell marker in carcinomas of head and neck, pancreas and breast. Survival rate is lower in CD44-positive patients as compared to those not expressing it [10]. This molecule designates a family of transmembrane glycoproteins responsible for cell adhesion. All CD44 isoforms are encoded by gene on chromosome 11 in humans. Exons 1-5 and 16-20 encode the constant position of all CD44 isoforms. Exons 6-15 encodes the variant exons are either completely excluded or are included in various isoforms combinations (CD44V). Exon 1 is not expressed in human cells due to presence of a stop codon. A positive link has been demonstrated in epithelial-mesenchymal transformation to generate CD44+ stem cells [11].
These cells were first identified in acute myeloid leukemia as a cell population expressing the surface antigen CD34+/CD38-. These cells possess the capacity to reproduce complete leukemic hierarchy following a xenograft. There are five defining criteria established to confirm cancer stem cells [12,13]. 1) Self-renewal capacity; 2) Restriction to a small number of total tumor cell population; 3) Reproducible tumor phenotype; 4) Multipotent differentiation to non-tumorigenic cells; and 5) Expression of distinct cell surface markers with persistent isolation [13].
Cancer stem cells are resistant to chemotherapy due to their ability to divide at a slower rate. A cancer stem cell population is characterized by three features:
a) Only a small number of cells within tumor population are associated with tumorigenic potential
b) Cancer stem cells are characterized by specific cell marker(s)
c) Tumors arising from these stem cells contain both tumorigenic and non-tumorigenic cell population
Human Telomerase Reverse Transcriptase (hTERT) expression is not detectable in normal tissues. Its expression in cancerous cells is the result of tumor-specific genetic and epigenetic events. Bmi1 gene, which is responsible for stem cell renewal upregulates hTERT expression in epithelial cells [12].
CONCEPT OF TUMOR HETEROGENEITY
‘Tumor heterogeneity’ represents differences among tumor cell phenotype and functions. Factors responsible for tumor heterogeneity are genomic heterogeneity, hierarchial tissue organization, environmental influences and mutations. The mutated tumor cells undergo Darwinian selection. These cells get influenced by microenvironmental influences exhibited by stromal cells and extracellular matrix. Cancer stem cells distinctly possess a feature of xenobiotic efflux due to an increased expression of membrane-bound proteins belonging to the ABC transporter family [2]. The cancer stem cell hypothesis assumes an hierarchial structure of cancer stem cells which is capable of self-renewal and heterogenous cell population production [2].
DRUG RESISTANCE IN CANCER STEM CELLS
Therapeutic approaches cannot eradicate the cancer stem cell compartment. This persistence is responsible for the disease relapse and [12]. Cancer stem cells acquire resistance towards chemotherapy due to multiple mechanisms which can be explained through many models [13].
Model 1 postulates that intrinsic mutations are harboured by a small cell population which confers property of drug resistance in these cells. Modes of acquiring mutations include increased ABC transporter expression, enhanced DNA repairing capacity and resistance towards apoptosis.
Model 2 suggests that cancer stem cell acquires genetic aberrations and mutations which confer drug resistance, thus, providing a selective advantage to these cells.
Model 3 suggests that drug-resistant variants of tumor stem cells produce multidrug resistant tumor cell variants, which exhibit quiescence, DNA repair ability and enhanced ABC transporter expression.
Model 4 is based on the concept that cancer stem cells accumulate genetic mutations on exposure to irradiation and carcinogens. These accumulated genetic alterations are transferred to the new generation of cancer stem cell.
According to the 5th model, the cancer stem cells and their progeny possess inherent drug resistance.
According to the 6th model, tumors possess a built-in drug-resistant pluripotent stem cell population which survives chemotherapy and are responsible for recurrence.
CANCER STEM CELLS-RELATED PROTEINS AND THEIR FUNCTIONS
Cancer stem cells are tumor-initiating resulting from malignant transformation of stem or progenitor cells. The cancer stem cells can possess stem cell-like properties such as self-renewal, proliferation and differentiation capabilities. Expressions of pluripotent markers such as- Sox2, Oct4, Nanog, CD133, ALDH1 and CD34+CD38- are evidences of stem cell-like nature. Cancer stem cells induce tumor vascularization by promoting neovascularization and angiogenesis via expression of factors such as- VEGF and PDGF. Tumor vascularization is a vicious process contributing to cancer progression as well as maintenance. Cancer stem cells acquire migratory properties through epithelial-mesenchymal transition. The adult stem cells are subjected to age-related alterations such as- telomeric DNA reduction, DNA repair defects, chromosomal rearrangements, genotoxic damage and accumulation of various genetic and/or epigenetic changes. The transformation of an adult stem cell into cancer stem cell is a long process and contributes to tumorigenesis and carcinogenesis. Telomeric alteration or gain of oncogenic events like- BCR-AML in hematopoietic stem cells allows selection of tumor cells resistant to senescence and apoptosis. Telomerase is expressed by adult and cancer stem cells. Telomerase expression is governed by an internal RNA subunit. The expression of telomerase enzyme decreases with age, resulting in telomeric shortening with increasing age. On becoming too short, p53-mediated DNA-damage response becomes activated that causes apoptosis. Downregulation of tumor suppressor gene, p53 in telomerase-deficient mice have been shown to induce carcinogenesis. Thus, the cancer stem cells generate from stem cells possessing short telomeres, which result in genomic instability and accumulation of mutations in oncogenes and tumor suppressor genes. Also, telomerase activity is increased in cancer stem cells resulting in tumorigenesis. Telomerase activity has been found to be reactivated in 85%-90% of cancers. However, on inhibition, tumor cells express a telomerase-independent system, termed as the ‘Alternative Lengthening of Telomeres (ALT)’. Inactivation of tumor suppressor genes such as- p53, PTEN and oncogene activation (e.g., Ras, Bmi1 and c-myc) play an important role in tumorigenesis. Activation of p53 in stem cells induces their apoptosis by stimulation of pro-apoptotic proteins such as Bak or downregulation of anti-apoptotic genes like- bcl-2 and bcl-xl. p53 downregulation can take place due to various factors such as- inactivation of p14ARF gene deletion or MDM2 gene multiplication. This downregulation of allows inhibition of apoptosis. Additionally, PTEN and PTEN/PI3/Akt have also been associated with carcinogenesis due to cancer stem cell stimulation. The Wnt/β-catenin pathway regulates the potency and differential potential of cancer stem cells. Abnormal activation of this pathway is related to the cancer stem cell maintenance and tumor development.
Upregulation of PLAGL-2, an oncoprotein promotes the self-renewal and proliferation capability of Cancer stem cells through Wnt/β-catenin pathway activation. Cancer epigenomes exhibit global DNA hypomethylation associated with hypermethylation at definitive promoters. DNA ‘hypomethylation’ leads to genomic instability due to chromosomal reorganization. Feinberg proposed the ‘Epigenetic Carcinogenesis Model’ for cancer stem cell origin [14,15]. As per this model, cancer stem cell can arise due to three steps: a) Epigenetic alterations; b) Mutation-mediated activation of an oncogene or silencing of a tumor suppressor gene; and c) Genetic and epigenetic instability. Evidence in favor of this hypothesis are: a) both embryonic and cancer stem cells share similar epigenetic profiles; and b) most cancers arise in elderly, thus, indicating epigenetic alterations in cancer stem cells [14,16].
Hans Clevers and his associates in the late 190s discovered Lgr5, a marker for tissue stem cells [17]. It is found to play an important role in dysregulated molecular signaling pathways in colon cancer. It is a cell surface receptor protein involved in the Wnt pathway. This pathway plays an important role in stem cell regulation and function as it involves APC or β-catenin proteins. Tcf-4 gene maintains the crypt stem cells within the small intestinal epithelium via the Wnt-β-catenin pathway. The APC-deficient human epithelial cells reflect upon the constitutive Tcf-4 activity, thus, contributing to malignant transformation. All Tcf-4 target genes are basically cancer genes. Lgr5 regulates the Wnt target gene expression. The Lgr5 expression is in turn enhanced by respondin, a stem cell growth [14] (Figure 1).
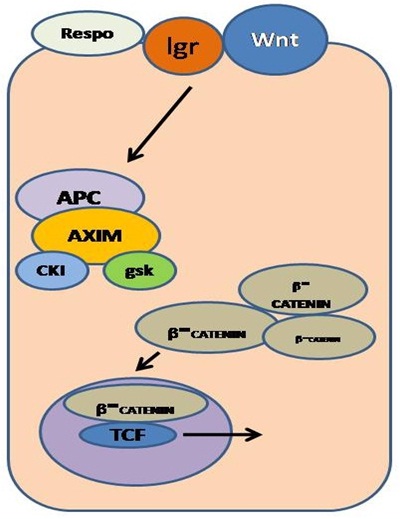
Figure 1: Regulatory WNT pathway in stem cell involving interaction with Lgr and Respondin proteins.
Proteome homeostasis or proteostasis is a mechanism for cell function, which regulates protein synthesis, localization and folding as well as removal of misfolded or aggregated protein. These damaged proteins can accumulate within cells with age, thus, instigating cell malfunction and death due to membrane damage. Lui et al., proposed a model to clarify the role of proteostasis in different stem cell types and their differentiated progeny [16,18]. Both of the stem and differentiated cells possess high-quality proteostasis, decreasing the effects caused by damages proteins, thus, allowing their long-term survival. Under these conditions, transcription factor ‘heat shock factor 1’ gets activated and induces HSPs expression which undertake folding of misfolded proteins preventing their aggregation. HSP90, a HSF1 target is a molecular chaperone which supports protein folding, thereby, preventing their aggregation. If the molecular chaperones cannot repair these damaged or misfolded proteins, they undergo degradation process through autophagy or proteosomes [16].
MAINTENANCE OF TUMOR CELLULAR POPULATION HIERARCHY
All tumors comprise of a heterogenous cell population. The ‘cancer stem cell’ hypothesis assumes that a tumor has hierarchial organization and only a small number of cells termed as ‘cancer stem cells’. These cells reside in a niche defined by microenvironmental conditions dictated by surrounding stromal cells; vasculature and extracellular matrix [16]. There are three mechanisms by which cancer stem cells replace older tumor cells: a) Genetic drift; b) Variations in growth rate of different clones; and c) Selection process.
a) Genetic drift: Deleterious mutations may accumulate in asexually dividing cell population due to lack of a mechanism for their elimination. For deleterious mutation, a random drift fixes a mutation, if U/S>1, where U=total number of mutation per genome; S=selection pressure against individual mutation. These deleterious mutations accumulate and result in random drift.
b) Variation in intrinsic rate of increase: Intrinsic rate of increase is defined as ‘the rate at which a cell population increases’. It can be described by the equation, dx/dt=rx where dx=change in number of cells with time interval; r=intrinsic rate of increase and x=population size at the beginning.
c) Selection: Hypoxia, immune response and therapeutic intervention allows for the selection of most adapted tumor cells in a population. This selection pressure on cancer cells preserves gene function which helps in their survival in variable ecological microenvironments or niches [16].
Tumor recurrence site is influenced by microenvironmental factors. Tumor cells’ adhesion to extracellular matrix activates signature genes, which play an important role in cancer initiation, progression or dormancy. Tumor dormancy can be described as a state of clinical remission wherein cancer cells remain occult for a long period of time. In head and neck cancers, increased Transforming Growth Factor-β2 (TGF-β2) has been found to be increased. Tumor cell dormancy is supported by Extracellular Matrix (ECM) remodeling, signaling of p38α, EGFβ2 and NR2F1 along with inhibition of c-jun, FAK, ERK1/2 and FOXM1 pathways. Cancer stem cells are heterogenous in nature and are broadly classified into three categories: a) Epithelial, b) those undergoing transition from epithelial to mesenchymal; and c) mesenchymal. ECM-positive cancer stem cells undergo genetic drift, clonal evolution and selection. Hence, this fact can be utilized to design personalized therapy to suppress the EMT-transformed cancer stem cells. Type II Transglutaminase (TG2) stimulates epidermal cancer stem cell epithelial-mesenchymal transition. This is a multifunctional enzyme, which acts as transamidase, GTP binding protein, protein scaffold, protein kinase and DNA hydrolase. TG2 is required for stem cell survival and maintenance in breast and ovarian carcinoma [19].
“Cancer clone evolution” occurs in tissues from wherein cancer develops by acquisition of mutations with time. The initial mutation termed as ‘gate-keeping mutation’ provides cells with selective growth advantage over normal cells. Examples of gate-keeping mutation involve the APC and KRAS genes. Natural selection occurs within a tumor through competition for space and nutrients. This selection process results in adaptive or phenotypic changes in cancer cells. Most cancer cells possess recurrent multiple mutations termed as ‘driver mutations’. These are specific mutations responsible for providing a selective growth advantage to a mutated cell. A driver gene comprises of both driver and non-driver mutations. Driver mutations truncate the encoded protein within its N-terminal. Non-driver mutations do not cause selective growth advantage in the cells. Passenger mutations do not affect initiation and progression of a tumor [19]. The ‘Oncogene addiction hypothesis’ suggests that tumor phenotype maintenance depends upon continuous oncogenic expression. “Inter-metastatic heterogeneity” is the difference between metastatic lesions in the same patient, while “intra-metastatic heterogeneity” refers to differences between cells of a metastatic tumor mass. The ‘lineage addiction hypothesis’ explains the involvement of dysregulated specific lineage pathways in tumor formation and progression. This model does not involve the acquisition of new and tumor-specific functions [20].
The ‘Hydra Model’ of cancer stem cell development explains the existence of numerous sub-clones. This sub-clone formation precedes the development of frank neoplasm. This hypothesis is consistent with the multi-hit theory of carcinogenesis [12].
The “Dynamic stem cell concept” suggests that the early stage cancers are governed by a single clone. The advanced stage tumors contain several distinctive clones, which arise either from initial cancer stem cell clone or from its differentiated cell progeny via various mutations or by induction of cancer stem cell niche. During carcinoma progression, cancer stem cell clones compete with each other leading to at least one cancer stem cell clone with loss of differentiation. With time, this process leads to reduction in hierarchial structure and leads to the selection of most aggressive cancer stem cells [11].
A cancer cell is a deviation of normal cell development closely controlled by factors present in embryonic microenvironment during cancer development. Injection of embryonic carcinomatous cells into a blastocyst cavity results in loss of their malignant behaviour. This microenvironmental influence is position-dependent as evident by persistence of malignant features when introduced within perivitelline space. Thus, either various differentiation factors present within the microenvironment variably interact with cancer cells and can cause differentiation or apoptosis [20]. “Stationary cancer stem cells” are present in epithelial tissues and are active in benign precursor lesions like adenoma. These cells persist in various differentiated states during tumor progression. The term “mobile cancer stem cells” describes stem cells located at the ‘tumor-host’ interface. The migrating cancer stem cells undergo epithelial-mesenchymal transition resulting in metastasis [11].
ROLE OF HEDGEHOG SIGNALING IN CANCER STEM CELLS
Abnormal Hedgehog signaling is associated with various human malignancies like- basal cell carcinoma, medulloblastoma, lung and pancreatic carcinomas. Constitutive pathways activate through loss-of-function mutations, epigenetic alterations and decreased expression of negative regulators like- PTCH, HHIP, SUFU, gain-of-function mutations and epigenetic influence on positive regulator, SMO. Hedgehog signaling has been found to play an important role in myeloid malignancies. The Hedgehog signaling pathway is dependent on three ligands- Sonic hedgehog, Indian hedgehog and Desert hedgehog. This gene is expressed during embryogenesis. The Hedgehog (Hh) ligands are synthesized as an inactive 45kDa precursor which undergoes post-translational modifications to form a 19kDa amino terminal active signaling molecule. The cholesterol and palmitoyl modifications are catalyzed by Hh acyl-transferase enzyme. This modification enhances the ligand activity and modifies its differentiation ability. The Hh ligands bind to the 12 transmembrane receptor proteins, PTCH1 (Patched 1). This results in its internalization and removes the Smoothened (SMO) repression. SMO causes accumulation of full length active form of zinc transcription factors, GLI-2 and GLI-3 within nucleus and also, potentiates activity of positive regulators of the pathway which includes serine threonine kinase 36 and kinisin family member 7, thus, resulting in transcription of downstream protein targets like GLI1 and PTCH1 which are regulators of chromatin formation, cell cycle activity, apoptosis and cellular mobility [11]. DISP, a 12-member transmembrane receptor protein is not involved in Hh ligand synthesis or processing but facilitates ligand movement [15].
CONCLUSION
Cancer stem cells are precursors of a specialized clone of cells bearing distinct genetic constitution than the normally regulated stem cells. A wide variety of mechanisms and their interrelationships govern their activity, metabolism and functions. Understanding these interrelationships is an important tool in combating their persistent nature and planning disease treatment.
REFERENCES
- Konstantinov IE (2000) The Man behind the Unitarian Theory of Hematopoiesis. Perspect Biol Med 43: 269-276.
- Fulawka L, Donizy P, Halon A (2014) Cancer stem cells-the current status of an old concept: literature review and clinical approaches. Biol Res 47: 66.
- Wicha MS, Liu S, Dontu G (2006) Cancer stem cells: an old idea--a paradigm shift. Cancer Res 66: 1883-1890.
- Cieply B, Koontz C, Frisch SM (2015) CD44S-hyaluronan interactions protect cells resulting from EMT against anoikis. Matrix Biol.
- Valent P, Bonnet D, Wöhrer S, Andreeff M, Copland M, et al. (2013) Heterogeneity of neoplastic stem cells: theoretical, functional, and clinical implications. Cancer Res 73: 1037-1045.
- Vermeulen L, Sprick MR, Kemper K, Stassi G, Medema JP (2008) Cancer stem cells--old concepts, new insights. Cell Death Differ 15: 947-958.
- Nowell PC (1989) The clonal nature of neoplasia. Cancer Cells 1: 29-30.
- Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, et al. (1988) Genetic alterations during colorectal tumor development. N Engl J Med 319: 525-532.
- Campbell LL, Polyak K (2007) Breast tumor heterogeneity: Cancer stem cells or clonal evolution? Cell Cycle 19: 2332-2338.
- Weismann IL, Anderson DJ, Gage F (2001) Stem and progenitor cells: origins, phenotypes, linear commitments and transdifferentiaition. Annu Rev Cell Dev Biol 17: 387-403.
- Orian-Rousseau V (2015) CD44 Acts as a Signaling Platform Controlling Tumor Progression and Metastasis. Front Immunol 6: 154.
- Topaloglu O, Sahin M, Delibasi T (2012) Basic tissues on cancer stem cells- concepts, in vitro models and therapeutic implications. Niche 1: 17-20.
- Styczynski J, Drewa T (2007) Leukemic stem cells: from metabolic pathways and signaling to a new concept of drug resistance targeting. Acta Biochim Pol 54: 717-726.
- Baccelli I, Trumpp A (2012) The evolving concept of cancer and metastasis stem cells. J Cell Biol 198: 281-293.
- Santos Franco S, Raveh-Amit H, Kobolák J, Alqahtani MH, Mobasheri A, et al. (2015) The crossroads between cancer stem cells and aging. BMC Cancer 15: 1.
- Clevers H (2013) The intestinal crypt, A prototype stem cell compartment. Cell 154: 274-284.
- Lui SK, Vilchez V, Gedaly R (2015) Liver Cancer Stem Cells: A New Paradigm for Hepatocellular Carcinoma Treatment. J Stem Cell Res Ther 5: 1-7.
- Korinek V, Barker N, Moerer P, van Donselaar E, Huls G, et al. (1998) Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat Genet 19: 379-383.
- Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T (2005) Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat Rev Cancer 5: 744-749.
- Feinberg AP, Ohlsson R, Henikoff S (2000). The epigenetic progenitor origin of human cancer. Nat Rev Genet 7: 21-35.
Citation: Citation: Chatterjee S, Damle SG, Sharma AK (2015) Cancer Stem Cells: Understanding their Dynamics. J Cancer Biol Treat 2: 005.
Copyright: © 2015 Shailja Chatterjee, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.