Distinctive Roles for the FOXO1 Transcription Factor in Immunological Development, Homeostasis and Malignancy
*Corresponding Author(s):
Luis MateronDepartment Of Biology, University Of Texas-Pan American, Texas, United States
Tel:9566657140,
Fax:9566653657
Email:lmateron@utpa.edu
Abstract
Forkhead box transcription factors of the O subclass: FOXO1, FOXO3, FOXO4 and FOXO6 impact diverse cellular processes in cell type specific manners. FOXO factors serve redundantly as tumor suppressors within the context of numerous cancers by hindering cell cycle progression and inducing apoptosis. However FOXO -1, -3,-4 and -6 homologs also possess distinct cellular roles as each knockout mouse has a unique phenotype. This review focuses on recent ground breaking discoveries that reveal FOXO1 has specific cellular roles in B cell development, T cell homeostasis as well as surprising, positive contributions to a number of immunological malignancies. These findings challenge conventional models that describe FOXO1 activities not only in the immune system, but in all cellular environments.
Keywords
TABLE OF CONTENTS
- Introduction
- Domain Structure and Regulation of FOXO1
- FOXO1:a Redundant Tumor Suppressor in Immune Cells
- FOXO1 in B Cell Specification and Homeostasis
- FOXO1 in T Cell Activation and Homeostasis
- FOXO1: the Surprising Putative Oncogene
- Conclusions and Future Directions
INTRODUCTION
Forkhead box transcription factors are characterized by a 100 amino acid winged helix DNA binding domain [1]. There are over 100 Forkhead box transcription factors found in organisms ranging from yeast to mammals [1,2]. Forkhead factors are subdivided into subclasses based in phylogeny [1]. The Forkhead box subclass O in mammals includes ubiquitously expressed homologs FOXO1, FOXO3, and FOXO4 as well as the FOXO6 homolog whose expression is predominantly in the brain [2,3]. Over expression in vitro studies with the FOXO -1, -3 and -4 homologs have shown that these factors have important, redundant roles in tumor suppression and stress response [2,4]. Triple Knock Out (TKO) murine models for these factors showed that they have partially redundant roles in tumor suppression [5,6]. However, additional murine loss of function models for these factors revealed that loss of each factor individually leads to distinct phenotypes [7-9]. The most strikingly loss of function phenotype was observed in Foxo1-/- mice, which are embryonic, lethal at day E10.5 with severe vascular defects [9]. Surprisingly, the loss of Foxo3 or Foxo4 produced progeny that were on a gross level similar to littermate controls. Foxo3-/- mice undergo premature ovarian failure and for many years Foxo4-/- mice lacked a discernible phenotype [9]. Tissue specific conditional knock out studies with Foxo1 have revealed very specific, novel roles for this factor in immunological processes [10]. This review will highlight how recent discoveries regarding Foxo1 in the field of immunology drastically revise conventional models for this factor.
DOMAIN STRUCTURE AND REGULATION OF FOXO1
The overall domain structure of FOXO1 includes an N-terminal Forkhead box DNA binding domain and a C-terminal region that promotes transcriptional activation, (Figure 1) [11]. Even though FOXO1 is primarily described as a transcriptional activator, its appreciated cellular roles are evolving [2]. There are instances when FOXO1 represses transcription such as the CCND2 gene encoding cyclinD2 [12]. Recently, FOXO1 has also been implicated in affecting the splicing of at least one gene, Ikaros in B cells [13]. ChIP seq. analysis of FOXO1 in B cells strikingly revealed that most of its binding sites are intragenic or intronic suggesting roles for FOXO1 that remain to be discovered [14]. FOXO1 impacts far reaching cellular processes in a cell type specific manner [5]. FOXO1 promotes gluconeogenesis, longevity and apoptosis in a manner that is directed by the integration of intracellular and extracellular cues in a highly context dependent manner [7]. Thus, FOXO1 has many cellular roles, but the structural determinants and responsible mechanisms that enable this molecule to perform these diverse roles remain to be delineated. The activity of FOXO1 (as well as related FOXO homologs) is heavily regulated at the post-transcriptional level. The most canonical regulatory mechanism for FOXO1 (and FOXOs -3 and -4), is phosphorylation on conserved residues by the AKT protein kinase [15,16]. This phosphorylation leads to the interaction of FOXO with the 14-3-3 scaffolding protein, leading to cytosolic localization/inactivation [15]. Therefore, elevated AKT activity leads to a diminishment in FOXO activity. SGK is also able to phosphorylate FOXO on these residues to hinder activity [17]. A host of other kinases also phosphorylate FOXO1 to modulate its activity including cyclin dependent kinase 1, which phosphorylates S249 and the Ste20-like kinase MST1 which phosphorylates FOXO1 on S207 leading to nuclear localization [18-20]. FOXO1 post-translational regulation is not restricted to phosphorylation events. This factor is also regulated by acetylation, arginylation, methylation and ubiquitination [21-26]. Much remains to be learned about how these regulatory events impact the ability of FOXO1 to perform its impressive array of cellular activities including those in the immune system.
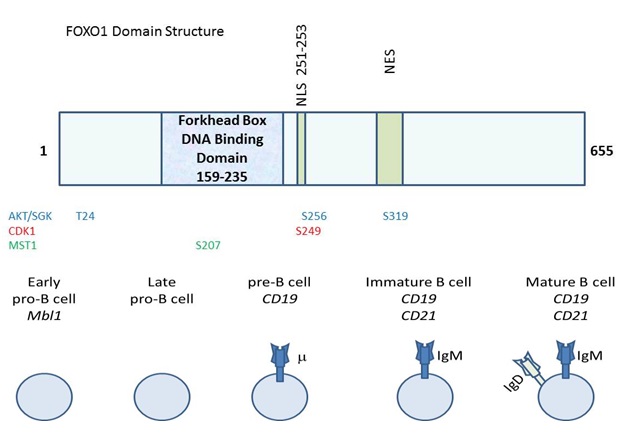
FOXO1: A REDUNDANT TUMOR SUPPRESSOR IN IMMUNE CELLS
One of the most commonly studied functions of FOXO1 (and FOXOs -3 and -4) is its ability to act as a tumor suppressor [5,27]. Initially, gain of function analyses led to the finding that FOXO1 induces the expression of genes that either hinder cell cycle progression or induce apoptosis in order to counter oncogenesis [28]. Exogenous FOXO1 induces the expression of CDKN1B (encodes p27) in primary mouse T cells and in CTLL-2 T-cells and cyclin G2 (CCNG2) as well as Rb p130 in primary mouse B cells to halt cell cycle progression [29-31]. To activate apoptosis, over-expression of FOXO1 induces the expression of Bim in CTLL-2 T cells and primary human T cells [30,32]. Exogenous FOXO1 also impacts proliferation in Jurkat T cells and primary human T cells [31,33]. FOXO1 is commonly expressed in Non-Hodgkin's lymphoma, but is frequently not expressed in classical Hodgkin's Lymphoma (cHL) [34]. In one study, FOXO1 expression was lost in 31 out of 32 primary classical Hodgkin's lymphomas (assessed by immuno-histochemistry), but was retained on other B cell lymphomas including follicular lymphoma, Diffuse Large B Cell Lymphoma (DLBCL) and MALTnon-Hodgkin's lymphoma [34]. Of note, classic Hodgkin's lymphoma has lost many B cell characteristics whereas other B cell lymphomas that retain FOXO1 expression also possess B cell characteristics. In line with this, FOXO1 has very specific, essential roles in B cell fate specification. [34,10]. Introduction of FOXO1 into 5 cHL cell lines induced cell cycle arrest and apoptosis supporting the idea that it is a tumor suppressor in this setting [34]. In addition to hematologic malignancies, an extensive body of research shows that FOXO1 acts as a tumor suppressor in a host of solid tumors such as breast cancer, prostate cancer, lung cancer and brain cancer in which it regulates an extensive array of genes to impact cellular processes including cell cycle progression and apoptosis [4,18,35-63]. Frequently, solid tumors and hematological malignancies have elevated PI3K and AKT, which leads to phosphorylation of FOXO inducing interaction with 14-3-3 scaffolding proteins and cytoplasmic localization [4,15]. Therefore, even if FOXO expression is retained in a cancer, elevated AKT activity is thought to inactivate its output.
A number of loss-of-function analyses corroborate the ability of FOXO factors to act as redundant tumor suppressors in immune cells.One murine model utilized Mx1-Cre which can be induced by polyinosine-polycytidylic acid (pI-pC) to obtain widespread somatic loss of Foxo1, Foxo3 and Foxo4 in mice at age 4-5 weeks [5]. Loss of all three FOXO factors led to the development of CD4+ CD8+ lymphoblastic lymphomas that spread to the spleen, liver and lymph nodes [5]. All six alleles of the FOXO factors needed to be lost in order for lymphoma to develop [5]. Even one FOXO allele could block lymphoma development highlighting the redundancy of these factors as tumor suppressors. The Cre+ mice also developed age-progressive hemangiomas that led to premature death [5]. In a similar study (using Mx1-Cre to knock out Foxo1, Foxo3 and Foxo4) it was found that loss of all three Foxos led to an increase in myeloid lineages including a two fold increase in white blood cells (attributable to an increase in neutrophils) as well as a 5.8 fold increase in reticulocytes [6]. Long term loss of Foxo led to a non-fatal myeloproliferative phenotype [6]. These mice also had aberrant B and T differentiation showing a significant decrease in mature B cells in the bone marrow and a slight block in T cell maturation at the DN1 to DN2 stage transition [6]. Strikingly, Foxo deficient HSCs showed abnormal cell cycle regulation, with an increase in cycling cells including a two fold increase in cells that were in S, G2 and M phases [6]. Concomitantly, Foxo target genes such as those encoding Cyclin G2, p130 Rb, p27 and p21 were down regulated with the loss of these factors whereas Cyclin D2 was increased. There were also significant increases in apoptosis and ROS in these cells [6]. Of note, the HSC restricted phenotypes (cell cycle alterations, apoptosis and ROS) could be suppressed by treatment of the Foxo deficient cells with the antioxidant N-acetyl-L-cysteine (NAC) [6]. This rescue implicates ROS as the mitigating factor that leads to cell cycle and apoptotic defects in HSCs upon Foxo loss and intriguingly hints that such mechanisms may explain Foxo phenotypes elsewhere. In addition to employing Foxodeletion mutants in murine cancer models, expression of dominant negative Foxo (contains the DNA biding domain of Foxo1, but lacks the transactivation domain) promotes lymphogenesis in an Eµ-Myc driven model [64]. In this study, hematopoetic stem cells that expressed Eµ-Myc developed lymphoma. Expression of dominant negative Foxoin this model greatly decreased the latency in lymphoma development by preventing the induction of 53-dependent apoptosis [64]. In sum, loss of all three Foxo factors in mice leads to a loss in the regulation of tumor suppressive outputs (such as proper induction of Cyclin G2 or p27) and lymphoma [5,6]. Additionally, these studies reveal that Foxo outputs reach beyond tumor suppression impacting immunological development and HSC maintenance [5,6].
FOXO1 IN B CELL SPECIFICATION AND HOMEOSTASIS
In contrast to the tumor suppressive role of FOXO1, which is redundant with the other Foxo transcription factors, its ability to impact B cell fate and maturation is highly specialized. Loss of Foxo1 during various stages during B cell development leads to a host of developmental and homeostatic deficiencies [10]. One of the major activities for Foxo1 in B-cells is to induce the expression of Rag1 and Rag2 [65,10]. Rag1 and Rag2 encode for recombinases that facilitate DNA rearrangements including heavy and light chain rearrangements during B cell development. Initial work in transformed AMuLV pro B cells showed that RNA interference targeting of Foxo1 (and not Foxo3 or Foxo4) led to a dramatic reduction in Rag1 and Rag2 expression [65]. Ectopic expression of Foxo1 (and not Foxo3 or Foxo4) promoted Rag1 and Rag2 expression in this setting. This study also found that PI3K inhibition increased Rag1 and Rag2 gene expression [65]. Interestingly loss of Foxo1 in thymocytes had no effect on Rag1 or Rag2 gene expression underscoring the cell type specific role of Foxo1 in regulating B cell gene expression [65].
Cell based studies strongly implied an important role for Foxo1 in B cell development. To examine this further, experiments were done in which Foxo1 was conditionally deleted during various stages of mouse development; see Table 1 [10]. Foxo1 was deleted using Cre driven by the Mbl1 promoter which targets expression during the early pro B cell stage of development. This led to a host of defects in B cell development and homeostasis. The spleen, lymph nodes and peripheral blood had 10% of the total number of B cells that were found in wild-type mice [10]. It was found that there was an increase in CD19-BP1- pre pro B cells, but decreases in early pro B cells (CD19+BP1-) and late pre B cells (CD19+BP1+), suggesting that B cell development was partially blocked at the pre pro B cell to pro B cell transition [10]. Subsequent analysis of this model revealed that there was a marked reduction in the expression of IL-7Ra, Rag1 and Rag2 expression. It was also found by chromatin immuno-precipitation analysis that Foxo1 associated with the ERAGenhancer [10]. Concomitantly, defects in DNA rearrangements were observed in this Rag deficient setting [10]. The Dengler study also employed additional conditional models to interrogate how Foxo1 impacts B cell development at later stages. CD19 promoter driven Cre led to partial loss of Foxo1 at the early pre B cell and immature B cell stages as well as nearly complete loss of Foxo1 at the mature B cell stage [10]. Loss of Foxo1 at this later stage led to defects in CD62L expression, which is critical for B cell homing to peripheral tissues. In line with this, there was a 30% reduction in lymph node B cells in this setting [10]. To examine Foxo1 loss during a later time point during B cell development the CD21 promoter driven Cre was utilized [10]. This allowed for the loss of Foxo1 after B cells had gone through IL7 dependent differentiation, IgG rearrangement and Rag mediated receptor editing [10]. This model revealed a number of intriguing phenotypes including lower expression of Aicda (encoding AID) leading to a reduction in class switch recombination [10]. Interestingly, there was also a reduction in phosphoAkt and phosphoErk upon BCR crosslinking in this model suggesting a positive role for Foxo1 in PI3K signaling within this context. Of note this study found that Foxo3 was not required for early B cell development, formation of IgG positive B cells and peripheral B cell differentiation, demonstrating that these roles are largely specific to Foxo1 [10].
| Murine model | Developmental stage | Phenotype | Study |
| Cre-Vav1Foxo1 -/- | Delete from hematopoetic compartment | Cells arrested at CLP LY6D+ stage, earlier B cell progenitor | Mansson et al. [66] |
| Cre-Mbl1Foxo1 -/- | Early lymphoid progenitor stage-earliest pro-B stage of development | Increased CD19-BP1- (pre pro B) Decreased CD19+BP1- (early pro B) Decreased CD19+BP1+ (late pre B) Blocked at pre pro B to early pro B |
Dengler et al. [10] |
| Cre-Mbl1 Foxo1 -/- | Early lymphoid progenitor stage-earliest pro-B stage of development | Failed to up-regulate IL-7Ra; cells were prone to apoptosis | Dengler et al. [10] |
| Cre-Mbl1 Foxo1 -/- | Early lymphoid progenitor stage-earliest pro-B stage of development | Decreased transcription of Rag1 and Rag2 with impaired VH to (D)JH rearrangement | Dengler et al. [10] |
| Cre-CD19 Foxo1 -/- | Partial deletion in pre B cells and immature B cells, nearly complete deletion by mature B cell stage | Decrease in VKJK rearrangement | Dengler et al. [10] |
| Cre-CD19 Foxo1 -/- | Partial deletion in pre B cells and immature B cells, nearly complete deletion by mature B cell stage | Increase in marginal zone B cells and decrease in follicular B cells Can suppress CD19- B cell deficiency |
Chen et al. [68] |
| Cre-CD21 Foxo1 -/- | Transitional stage of B ell development (after IL7 dependent differentiation, IgG rearrangement and RAG mediated receptor editing) | 30% reduction in lymph node B cells Reduced CD62L expression |
Dengler et al. [10] |
| Cre-CD21 Foxo1 -/- | Transitional stage of B cell development (after IL7 dependent differentiation, IgG rearrangement and RAG mediated receptor editing) | Survival defect specific to BCR crosslinking (not stimulation with IL4 or LPS) Decrease in IgM dependent Phospho-Akt and Phospho-Erk |
Dengler et al. [10] |
| Cre-CD21 Foxo1 -/- | Transitional B cell stage | Impaired class switch recombination (CSR) and Aicda (AID) expression | Dengler et al. [10] |
Once Foxo1 was established as being centrally important to B cell development subsequent investigations focused on delineating regulatory mechanisms for this factor and determining the stages during B cell development that are impacted by this factor. To investigate earlier stages of hematopoiesis, Cre placed under the regulation of the Vav1 promoter was utilized to deplete Foxo1 from the hematopoietic compartment. Foxo1 depleted mice had grossly normal BM cellularity with a two-fold reduction in splenic cells [66]. The number of BM CD19+B220+ cells was reduced by 50 fold with few CD19+ cells detected in the spleen [66]. The few CD19+ BM cells lacked expression of IgM or IgD suggesting that development was blocked at the pro B cell stage [66]. The CLP compartment (common lymphoid progenitor compartment) can be divided into LY6D- and LY6D+ forms. The LY6D- cells retain full in vivo lymphoid potential whereas LY6D+ have up-regulated B cell specifying factors such as EBF1 and Pax5 and act as B cell biased lymphoid progenitors [66]. Of note, Foxo1 depleted cells had a two fold increase in LY6D+ cells in the CLP compartment and remained arrested at this stage of development [66]. These cells showed an increase in cycling during the cell cycle and a decrease in apoptosis, consistent with previously ascribed functions for Foxo1 [67]. Mechanistically, Foxo1 was found to increase levels of EBF1 expression during early B cell development leading to a positive feedback loop in which both factors activated the expression of the other to specify B cell fate [66]. This work demonstrates that Foxo1 is needed in LY6D+ CLP cells to ensure proper B cell development [66].
Foxo1 is essential for proper B cell development from the early B cell biased lymphoid stage (LY6D+) through pro B cell, pre B cell, immature B cell and mature B cell stages. Foxo1 has also been recently identified as being essential in directing the proper balance between mature B cell lineages [68]. A lack of Foxo1 directed by CD19 Cre led to a loss in follicular B cells and a gain in marginal zone B cells [68]. CD62L and KLF2 mRNAs were reduced in Foxo1-/- B cells. Interestingly, CD19 deficient mice, which have lower PI3K activity, have greatly reduced marginal zone B cells [69]. Loss of the tumor suppressor Pten (performs the converse reaction to PI3K), suppresses the marginal zone defect that is found in the CD19 deficient background [70]. Likewise, the loss of Foxo1restores marginal zone B cellsto CD19 deficient mice [68]. In summary, true to conventional models of the PI3K pathway, high CD19/PI3K activity is required for marginal zone B cell development, whereas Pten and Foxo1 activity favor follicular B cell formation.
Over the last few years key regulatory events have been delineated that direct Foxo1 expression during B cell development. Based on mouse models, the earliest point that requires Foxo1 is in the LY6D+ CLP stage [66]. E2A and HEB act in concert to orchestrate the transition from CLP LY6D- to LY6D+ [71]. Mice that are E2A+/- and HEB -/- (utilizing Cre driven by the Tie2 promoter, deleting these factors from the entire hematopoietic system) are blocked as LY6D- CLP cells and fail to activate Foxo1 among other genes that have roles in B cell fate specification [71]. Additional mechanisms are employed to regulate FOXO1 expression during later stages of B cell development. For example, during the pro B cell stage of development E2A and EBF1 associate with the Foxo1 locus to induce expression [72]. This leads to the activation of an interlocked cis regulatory network that regulates B cell fate. Chromatin immuno-precipitation analyses with pro B cells (Rag1 deficient), showed that E2A, EBF1 and Foxo1 all predominantly associated with intronic and intragenic sequences [72]. Of the genes that were co-regulated by E2A and EBF1, a significant number was also enriched for Foxo1 association (p=9.88 × 10-324) [72]. EBF1 might not induce all genes that are co-regulated with Foxo1, as it appears that EBF1 represses the expression of an important Foxo1 target Rag1 [73]. EBF1 hinders Foxo1 recruitment to the Rag1 locus [73]. Of note, EBF1 also represses Foxo1 gene expression in large cycling pre B cells to allow for a brief period of proliferation [73]. Overall, numerous mechanisms are utilized to regulate Foxo1 gene expression during B cell development.
In addition to the regulation of Foxo1 at the transcriptional level during B cell development, Foxo1 is also regulated on the post-translational level by phosphorylation and protein-protein interactions. Chief among these events is the phosphorylation of Foxo1 by Akt on conserved residues leading to cytoplasmic sequestration/inactivation [31]. During periods of B cell proliferation, Foxo transcription factors must be inactivated to alleviate cell cycle arrest [31]. Using retroviral transduction vectors, exogenous expression of mutant Foxo1 and Foxo3 (have mutated residues to prevent Akt phosphorylation) led to increased cell cycle arrest and apoptosis [31]. These results are corroborated using Irf4-/-, Irf8-/- pre B cells as a model for large cycling B cells to study dynamics between Foxo1 and PI3K signaling [14]. It was found that IL7 kept pre B cells in a cycling proliferative state similar to what is observed during the limited clonal expansion of pre B cells during development [14]. Small and large B cells were isolated from the Irf4-/-Irf8-/- mice. It was found that the small cells had elevated levels of Rag gene expression [14]. Removal of IL7 from the media led to Foxo1 nuclear localization and recruitment to target genes such as ERAG1 and ERAG2 [14]. This work showed that IL7 activated PI3K to promote a brief period of clonal expansion followed by cell cycle arrest and differentiation [14]. Within an organismal context it was postulated that B cells may receive IL7 from the stroma and as cells asymmetrically divide away, the IL7 signal becomes less thereby triggering the activation of Foxo1 and cell cycle arrest. ChIP seq. analysis was performed to identify direct Foxo1 target genes in pre B cells from Irf4-/-Irf8-/- mice. Interestingly, 88% of the Foxo1 binding sites were either intergenic or intronic [14]. Foxo1 binding peaks were found near Rag1, Rag2, Blnk, and Syk. Syk is a tyrosine kinase that phosphorylates Blnk, which then interacts with BCR signaling molecules such as Grb2, Vav and Nck via their SH2 domains to promote downstream signal transduction. Subsequent work showed that Foxo1 and Pax5 physically associated with each other by co-immuno-precipitation analysis and that they co-regulated the Blnk gene upon IL7 removal [14]. The Foxo1 and Pax5 cistromes shared a 51% overlap suggesting that these factors commonly co-regulate genes [14]. The induction of Blnk by Foxo1 and Pax5 initiates a feedback loop as Blnk leads to lower Akt activity and therefore higher Foxo1 activity leading to sustained Blnk expression [14]. In this manner Foxo1 initiates positive feedback loops to maintain B cell specific programs.
Murine models have also investigated the regulation of FOXO1 by 14-3-3 scaffolding proteins. FOXO1 is phosphorylated on conserved residues by AKT, which greatly facilitates interaction with 14-3-3 scaffolding proteins and cytoplasmic sequestration [15]. The impact of 14-3-3 interaction on Foxo1 was interrogated using 14-3-3 knock-out mice obtained by conventional gene targeting [74]. The 14-3-3 deficient mice had only 1/3 of the wild-type number of splenic B cells [74]. Both marginal zone and follicular B cells were decreased in the 14-3-3 deficient model [74]. Interestingly, loss of 14-3-3 increased apoptosis and proliferation in B220+ B cells, suggesting an increase in Foxo function. Increased expression of numerous Foxo1 target genes was found in 14-3-3 deficient B cells (BIM and p27) along with a severe decrease in Foxo1 protein expression [74]. Therefore, loss of 14-3-3 leads to activated Foxo1, which may be less stable. Foxo1 is known to induce AKT S473 phosphorylation by up regulating the expression of Rictor, a key component of the mTORC2 complex that phosphorylates AKT on S473 [74]. However, in the 14-3-3 KO B cell setting there was a striking decrease in phospho AKT S473 suggesting that even though Foxo1 may be active, it fails to induce Rictor in this context [74]. Of note, there was a concomitant induction in mTORC1 activity in 14-3-3 KO cells as evidenced by an increase in phospho-S6 phosphorylation on S240/244 [74]. These data implied that 14-3-3 may be required for Foxo1 to properly regulate AKT activity via mTORC2. It was proposed that 14-3-3 may be required to maintain an elevated level of Foxo1 protein, which may be necessary for mTORC2 activation via Rictor induction. This appears to be the case as treatment of B cells with the proteasomal inhibitor MG132 stabilized Foxo1 protein expression and suppressed the induction of P-S6 S240/244 [74]. Thus, interaction between Foxo1 and 14-3-3 regulates Foxo1 activity and stability.
Foxo1 canonically regulates gene expression. However, a recent study indicates that Foxo1 also regulates Ikaros mRNA splicing to allow for Ig gene rearrangements [13]. Ikaros encodes for a Kruppel-like zinc finger transcription factor that facilitates the induction of Rag genes and accessibility of the IgH locus for recombination [75]. Foxo1 was found to be essential for Ikaros expression in primary B cells [13]. Mechanistically, Foxo1 was needed for proper splicing of Ikaros transcripts in pro B cells [13]. Instead of normal transcripts, pro B cells deficient for Foxo1 produced Ikaros transcripts that lacked exons 5 and 6 and had a 378 nucleotide insertion in exon 7 [13]. The mechanism for this aberrant splicing remains to be elucidated. It may be possible that Foxo1 directly regulates splicing by binding to intronic regions. It will be illuminating to examine whether Foxo1 impacts the splicing of other genes.
In summary a host of studies indicate that Foxo1 is essential in B cell development from the earliest stages of B cell lineage to the circulation of mature B cells [10]. Unlike tumor suppressive roles for Foxo1, which are largely redundant with other Foxo transcription factors, loss of Foxo1 alone during B cell development leads to severe defects including a block at the pre pro B cell to pro B cell stage and impaired class switch recombination [5,6,10].
FOXO1 IN T CELL ACTIVATION AND HOMEOSTASIS
In comparison to B cell development, Foxo1 has a minor impact on T cell development [76,77]. Foxo1 is required for naïve T cell homeostasis and T cell homing [78]. Initially the role of Foxo1 in regulating T cells was examined using exogenous expression of this factor in Jurkat cells (leukemic T lymphocytes) [79]. It was found that FOXO1 could strongly induce T cell homing genes such as CD62L, EDG6, EDG1, KLF2 and CCR7 [79]. Of note, the FOXO1 mutant H215R, which lacks histidine 215 that makes a key DNA contact led to decreased CD62L expression, suggesting that different forms of Foxo1 can induce different subsets of its genes in T cells [79]. FOXO1 was also found to repress the expression of CD69, known to be involved in early T cell activation [79]. Subsequent mouse models deleted Foxo1 from T cells using either Cre driven by the CD4 promoter or the Lck promoter. Mice obtained using CD4 Cre demonstrated that Foxo1 was nearly dispensable for T cell development [78,80]. Foxo1 deletion from T cells using Cre driven by the Lck promoter (induced at a slightly earlier stage double negative, CD4-CD8-, stage of T cell development) led to modest defects in T cell development [78]. These mice had slightly lower double positive thymocytes with concomitant increases in CD4+ and CD8+ single positive thymocytes [78]. The spleens and lymph nodes of the Foxo1 KO mice were smaller and contained half of the T cells that were found in WT controls [81]. The Foxo1 KO T cells had low CD62L and CCR7 [78,80]. The Klf2 gene was also reduced in Foxo1 KO T cells [81,82]. Purified Foxo1 KO lymph node T cells were defective in homing to lymph nodes when transplanted into lethally irradiated CD45.2+ mice demonstrating a homing defect [80]. Of note, the Foxo1 KO T cells were preferentially found in the spleen compared to wild type controls. There was no difference in the number of T cells found in the blood between Foxo1 KO mice and controls. Foxo1 was also found to be critical for the expression of Bcl2 in CD44lo CD4+ CD8+ T cells highlighting a potential role for Foxo1 in regulating survival of these cells [80]. Consistent with this notion, Foxo1 was also shown to bind to and activate IL7R-a gene expression in T cells [78]. Similar to B cells IL7, induces growth and survival programs via PI3K and can down regulate Foxo activity via phosphorylation by AKT in T cells as well [78].
Foxo1 is also a key regulator of T cell activation. Naïve T cells have already differentiated in bone marrow and have gone through positive and negative processes in the thymus, but have not come into contact with antigen in the periphery [82]. Naïve T cells lack markers of activation such as CD44 or CD69 [82]. Foxo1 KO mice generated with CD4 promoter regulated Cre had a higher percentage of CD44hi CD62Llo or CD69+cells in double positive T cells isolated from spleens [82]. The elevation in CD44 and CD69 positivity indicated that there may be a defect in maintaining naïve T cells in Foxo KO T cells. Consistent with these results, genes that are positive regulators of T cell activation Gadd45g, Tnfrsf9 and Rora were increased in Foxo KO T cells [83]. The mechanism by which Foxo1 hinders the expression of these genes remains to be determined.
Effector T cells can eventually differentiate into memory T cells [83]. The loss of Foxo1 using CD4 Cre in T cells leads to defects in memory T cell maintenance and function [83]. CD8+ memory T cells with Foxo KO develop in normal amounts, but over a period of time (30-95 days post infection) the Foxo KO memory T cells are reduced in number by an order of magnitude [83]. When mice are challenged with the Armstrong strain of LCMV following acute infection (45-60 days post-initial infection), the Foxo1 mutants were highly deficient in mounting a secondary response showing a defect in protective immunity post infection [83]. Effector T cells undergo a transition to form long lasting memory T cells. Part of this transition involves the down regulation of genes that enable effector cell function such as TBX21, encoding a transcription factor that induces INF-? expression, whereas other genes are required for the long term survival of memory T cells such as Eomes [84]. Foxo1 KO CD8+ memory T cells fail to fully transition to the memory T cell phenotype [84]. For example, Foxo1 KO CD8+ T cells maintained elevated levels of the effector specific factor TBX21 and failed to maintain high levels of Eomes [84]. CD4+ memory T cells also showed a defect with Foxo1 KO. These cells develop relatively normally, but fail to produce high levels of the cytokines TNF-a, IL2 and INF-? post infection. These results show that Foxo1 is important to memory T cell maintenance and function.
T cell responses can be suppressed by regulatory T cells called Tregs [85]. Regulatory T cells come in many forms including the commonly studied CD4+CD25+Foxp3+set [86]. Tregs serve an important immunological role by suppressing responses following the resolution of an infection and by suppressing autoimmune disease [86]. Foxo1 has been shown to activate the expression of Foxp3 to facilitate the development of Tregs upon hydrogen peroxide treatment as well as activation by Mst1 [19]. Future work in Foxo KO models with regard to Treg development and function will shed light on the specific role of this factor in Treg cell biology.
Overall, the impact of Foxo1 on T cell development is relatively minor compared to its central role in B cell development, (Table 20). In T cells, Foxo1 has important roles in homing and in maintenance of memory T cells [78,83].
FOXO1: THE SURPRISING PUTATIVE ONCOGENE
The role of FOXO transcription factors in cancer is constantly evolving. Recent discoveries highlight pro-oncogenic roles for these factors in a number of hematological malignancies [87,88]. First using a murine model for Acute Myeloid Leukemia (AML) that was driven by the MLL-AF9 fusion gene, the role of Foxo factors was tested [87]. All of the control mice succumbed to AML within 60 days, whereas 20% of Foxo triple knock-out mice (lacking Foxo1, Foxo3 and Foxo4) were disease free for up to 5 months post excision [87], (Table 2). Examination of leukemia development in this system revealed that the control mice had 16 fold more Leukemia Initiating Cells (LIC) compared to Foxo triple knock out models [87]. LICs are a small population of AML cells that can establish leukemia in recipient mice [87]. The biology that allows Foxo to promote leukemia remains to be completely delineated, but will likely identify novel roles for these factors in immune cells.
Given its central role in B cell development perhaps it is not too surprising that Foxo1 appears to be a proto-oncogene in Diffuse Large B Cell Lymphoma (DLBCL) [88]. Foxo1 is mutated to a presumably active state in 8.6% of DLBCL [88]. These mutations are highly associated with poor prognosis based on Kaplan Meier Survival analysis [88]. The Foxo1 mutations encode for changes that cluster to an N-terminal region near one of the amino acids that is phosphorylated by AKT (Threonine 28). These mutations abrogate T28 phosphorylation leading to increased nuclear localization of this factor [88], (Table 2). Additional mutants were found near the DNA binding domain of Foxo1, but do not alter amino acids that make direct contact with the DNA [11,88]. More rigorous analyses are needed to investigate exactly how Foxo1 mutations impact the development of DLBCL and whether this mechanism is found in other cancers.
| FOXO1 Mutant Phenotype | Source |
| T cell homeostasis defect in Foxo1 deletion mutant (CD4 or Lck driven Cre) | Ouyang et al. [78]; Kerdiles et al. [80] |
| T cell homing defect in Foxo1 deletion mutant (CD4 or Lck driven Cre) | Ouyang et al. [78]; Kerdiles et al. [80] |
| Aberrant T cell activation in Foxo1 deletion mutant (CD4 driven Cre) | Kerdiles et al. [80] |
| Memory T cell maintenance and function defect in Foxo1 deletion (CD4 driven Cre) | Tejera et al. [83]; Hess Michelini et al. [84] |
| Hinders AML development (MLL-AF9 transgenic model with conditional triple deletion of Foxo1, Foxo3 and Foxo4) | Sykes et al. [87] |
| DLBCL N-terminal FOXO1 mutants lead to increased nuclear localization | Trinh et al. [88] |
CONCLUSIONS AND FUTURE DIRECTIONS
Traditionally Foxo1 was thought to be largely redundant with homologs Foxo3 and Foxo4 [2]. However, conventional and conditional mouse models indicate that Foxo1 specifically impacts many developmental processes from the vasculature to the immune system [9,10]. Foxo1 is essential for B cell development from the CLP LY6D+ stage (early B cell biased lymphoid stage) to the development of and maintenance of mature B cells [10,66]. One of the key Foxo1 regulated events during B cell development is the induction of Rag1 and 2 recombinases that are required for DNA rearrangements during development [10,65]. Foxo1 also serves to home peripheral T cells in part through its induction of CD62L [68]. Finally, Foxo1 may have oncogenic roles in hematological malignancies. The targets for mutated Foxo1 in DLBCL remain to be identified, but these targets will likely have some overlap with identified targets for Foxo1 during B cell development. Another intriguing area for future research is delineating the functional consequences of Foxo1 binding to intronic and intergenic sequences. Does this binding facilitate the regulation of chromatin structure and/or splicing? The precise role of Foxo1 in Treg cells remains an open area of investigation. In sum, recent studies have completely changed our view Foxo1 as a redundant tumor suppressor. Groundbreaking work revealed that Foxo1 has a very specific impact on immunological development and maintenance and surprising roles as aputative oncogene in DLBCL.
ACKNOWLEDGMENTS
The authors would like to thank members of the Keniry and Materon laboratories for helpful discussions.
REFERENCES
- Kaestner KH, Knochel W, Martinez DE (2000) Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev 14: 142-146.
- Calnan DR, Brunet A (2008) The FoxO code. Oncogene 27: 2276-2288.
- Jacobs FM, van der Heide LP, Wijchers PJ, Burbach JP, Hoekman MF, et al. (2003) FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. J Biol Chem 278: 35959-35967.
- Keniry M, Parsons R (2008) The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene 27: 5477-5485.
- Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, et al. (2007) FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 128: 309-323.
- Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, et al. (2007) FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 128: 325-339.
- Accili D, Arden KC (2004) FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 117: 421-426.
- Arden KC (2008) FOXO animal models reveal a variety of diverse roles for FOXO transcription factors. Oncogene 27: 2345-2350.
- Hosaka T, Biggs WH, Tieu D, Boyer AD, Varki NM, et al. (2004) Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc Natl Acad Sci U S A 101: 2975-2980.
- Dengler HS, Baracho GV, Omori SA, Bruckner S, Arden KC, et al. (2008) Distinct functions for the transcription factor Foxo1 at various stages of B cell differentiation. Nat Immunol 9: 1388-1398.
- Brent MM, Anand R, Marmorstein R (2008) Structural basis for DNA recognition by FoxO1 and its regulation by posttranslational modification. Structure 16: 1407-1416.
- Schmidt M, Fernandez de Mattos S, van der Horst A, Klompmaker R, Kops GJ, et al. (2002) Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol Cell Biol 22: 7842-7852.
- Alkhatib A, Werner M, Hug E, Herzog S, Eschbach C, et al. (2012) FoxO1 induces Ikaros splicing to promote immunoglobulin gene recombination. J Exp Med 209: 395-406.
- Ochiai K, Maienschein-Cline M, Mandal M, Triggs JR, Bertolino E, et al. (2012) A self-reinforcing regulatory network triggered by limiting IL-7 activates pre-BCR signaling and differentiation. Nat Immunol 13: 300-307.
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, et al. (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96: 857-868.
- Manning BD, Cantley LC (2007) AKT/PKB signaling: navigating downstream. Cell 129: 1261-1274.
- Brunet A, Park J, Tran H, Hu LS, Hemmings BA, et al. (2001) Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol Cell Biol 21: 952-965.
- Choi HK, Cho KB, Phuong NT, Han CY, Han HK, et al. (2013) SIRT1-mediated FoxO1 deacetylation is essential for multidrug resistance-associated protein 2 expression in tamoxifen-resistant breast cancer cells. Mol Pharm 10: 2517-2527.
- Du X, Shi H, Li J, Dong Y, Liang J, et al. (2014) Mst1/Mst2 regulate development and function of regulatory T cells through modulation of Foxo1/Foxo3 stability in autoimmune disease. J Immunol 192:1525-1535.
- Yuan Z, Becker EB, Merlo P, Yamada T, DiBacco S, et al. (2008) Activation of FOXO1 by Cdk1 in cycling cells and postmitotic neurons. Science 319: 1665-1668.
- Frescas D, Valenti L, Accili D (2005) Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem 280: 20589-20595.
- Matsuzaki H, Daitoku H, Hatta M, Aoyama H, Yoshimochi K, et al. (2005) Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc Natl Acad Sci U S A 102: 11278-11283.
- van der Heide LP, Smidt MP (2005) Regulation of FoxO activity by CBP/p300-mediated acetylation. Trends Biochem Sci 30: 81-86.
- Vogt PK, Jiang H, Aoki M (2005) Triple layer control: phosphorylation, acetylation and ubiquitination of FOXO proteins. Cell Cycle 4: 908-913.
- Yamagata K, Daitoku H, Takahashi Y, Namiki K, Hisatake K, et al. (2008) Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol Cell 32: 221-231.
- Yang Y, Zhao Y, Liao W, Yang J, Wu L, et al. (2009) Acetylation of FoxO1 activates Bim expression to induce apoptosis in response to histone deacetylase inhibitor depsipeptide treatment. Neoplasia 11: 313-324.
- Huang H, Tindall DJ (2006) FOXO factors: a matter of life and death. Future Oncol 2: 83-89.
- Nakamura N, Ramaswamy S, Vazquez F, Signoretti S, Loda M, Sellers WR et al. (2000) Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Mol cell biol 20: 8969-8982.
- Chen J, Yusuf I, Andersen HM, Fruman DA (2006) FOXO transcription factors cooperate with delta EF1 to activate growth suppressive genes in B lymphocytes. J Immunol 176: 2711-2721.
- Stahl M, Dijkers PF, Kops GJ, Lens SM, Coffer PJ, et al. (2002) The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J Immunol 168: 5024-5031.
- Yusuf I, Zhu X, Kharas MG, Chen J, Fruman DA (2004) Optimal B-cell proliferation requires phosphoinositide 3-kinase-dependent inactivation of FOXO transcription factors. Blood 104: 784-787.
- Bosque A, Aguiló JI, Alava MA, Paz-Artal E, Naval J, et al. (2007) The induction of Bim expression in human T-cell blasts is dependent on nonapoptotic Fas/CD95 signaling. Blood 109: 1627-1635.
- Fabre S, Lang V, Harriague J, Jobart A, Unterman TG, et al. (2005) Stable activation of phosphatidylinositol 3-kinase in the T cell immunological synapse stimulates Akt signaling to FoxO1 nuclear exclusion and cell growth control. J Immunol 174: 4161-4171.
- Xie L, Ushmorov A, Leithäuser F, Guan H, Steidl C, et al. (2012) FOXO1 is a tumor suppressor in classical Hodgkin lymphoma. Blood 119: 3503-3511.
- Abdelnour-Berchtold E, Cerantola Y, Roulin D, Dormond-Meuwly A, Demartines N, et al. (2010) Rapamycin-mediated FOXO1 inactivation reduces the anticancer efficacy of rapamycin. Anticancer Res 30: 799-804.
- Brosens JJ, Lam EW (2013) Progesterone and FOXO1 signaling: harnessing cellular senescence for the treatment of ovarian cancer. Cell Cycle 12: 1660-1661.
- Chen Z, Xiao Y, Zhang J, Li J, Liu Y, et al. (2011) Transcription factors E2A, FOXO1 and FOXP1 regulate recombination activating gene expression in cancer cells. PLoS One 6: e20475.
- Feng X, Wu Z, Wu Y, Hankey W, Prior TW, et al. (2011) Cdc25A regulates matrix metalloprotease 1 through Foxo1 and mediates metastasis of breast cancer cells. Mol Cell Biol 31: 3457-3471.
- Goto T, Takano M, Hirata J, Tsuda H (2008) The involvement of FOXO1 in cytotoxic stress and drug-resistance induced by paclitaxel in ovarian cancers. Br J Cancer 98: 1068-1075.
- Guttilla IK, White BA (2009) Coordinate regulation of FOXO1 by miR-27a, miR-96, and miR-182 in breast cancer cells. J Biol Chem 284: 23204-23216.
- Han CY, Cho KB, Choi HS, Han HK, Kang KW (2008) Role of FoxO1 activation in MDR1 expression in adriamycin-resistant breast cancer cells. Carcinogenesis 29: 1837-1844.
- Hou T, Ou J, Zhao X, Huang X, Huang Y, et al. (2014) MicroRNA-196a promotes cervical cancer proliferation through the regulation of FOXO1 and p27Kip1. Br J Cancer 110: 1260-1268.
- Huang CY, Chan CY, Chou IT, Lien CH, Hung HC, et al. (2013) Quercetin induces growth arrest through activation of FOXO1 transcription factor in EGFR-overexpressing oral cancer cells. J Nutr Biochem. 24:1596-1603.
- Kim SY, Yoon J, Ko YS, Chang MS, Park JW, et al. (2011) Constitutive phosphorylation of the FOXO1 transcription factor in gastric cancer cells correlates with microvessel area and the expressions of angiogenesis-related molecules. BMC Cancer 11: 264.
- Lee SH, Kang YJ, Sung B, Kim DH, Lim HS, et al. (2014) MHY-449, a novel dihydrobenzofuro[4,5-b][1,8] naphthyridin-6-one derivative, induces apoptotic cell death through modulation of Akt/FoxO1 and ERK signaling in PC3 human prostate cancer cells. Int J Oncol 44: 905-911.
- Li Y, Yu J, DU D, Fu S, Chen Y, et al. (2013) Involvement of post-transcriptional regulation of FOXO1 by HuR in 5-FU-induced apoptosis in breast cancer cells. Oncol Lett 6: 156-160.
- Liu P, Li S, Gan L, Kao TP, Huang H (2008) A transcription-independent function of FOXO1 in inhibition of androgen-independent activation of the androgen receptor in prostate cancer cells. Cancer Res 68: 10290-10299.
- Lu H, Liu P, Pan Y, Huang H (2011) Inhibition of cyclin-dependent kinase phosphorylation of FOXO1 and prostate cancer cell growth by a peptide derived from FOXO1. Neoplasia 13: 854-863.
- Maekawa T, Maniwa Y, Doi T, Nishio W, Yoshimura M, et al. (2009) Expression and localization of FOXO1 in non-small cell lung cancer. Oncol Rep 22: 57-64.
- Myatt SS, Wang J, Monteiro LJ, Christian M, Ho KK, et al. (2010) Definition of microRNAs that repress expression of the tumor suppressor gene FOXO1 in endometrial cancer. Cancer Res 70: 367-377.
- Park J, Ko YS, Yoon J, Kim MA, Park JW, et al. (2014) The forkhead transcription factor FOXO1 mediates cisplatin resistance in gastric cancer cells by activating phosphoinositide 3-kinase/Akt pathway. Gastric Cancer 17: 423-430.
- Pei J, Lou Y, Zhong R, Han B (2014) MMP9 activation triggered by epidermal growth factor induced FoxO1 nuclear exclusion in non-small cell lung cancer. Tumour biol 35: 6673-6678.
- Wang X, Lin C, Zhao X, Liu A, Zhu J, et al. (2014) Acylglycerol kinase promotes cell proliferation and tumorigenicity in breast cancer via suppression of the FOXO1 transcription factor. Mol Cancer. 13:106.
- Wu Y, Elshimali Y, Sarkissyan M, Mohamed H, Clayton S, et al. (2012) Expression of FOXO1 is associated with GATA3 and Annexin-1 and predicts disease-free survival in breast cancer. Am J Cancer Res 2: 104-115.
- Yu DA, Yoon J, Ko YS, Park J, Kim SY, et al. (2014) Forkhead transcription factor FOXO1 inhibits nuclear factor-kappaB in gastric cancer. APMIS.
- Yu F, Jin L, Yang G, Ji L, Wang F, et al. (2014) Post-transcriptional repression of FOXO1 by QKI results in low levels of FOXO1 expression in breast cancer cells. Oncol Rep 31: 1459-1465.
- Yu JJ, Wu YX, Zhao FJ, Xia SJ (2014) miR-96 promotes cell proliferation and clonogenicity by down-regulating of FOXO1 in prostate cancer cells. Med Oncol 31: 910.
- Zeng Z, Lin H, Zhao X, Liu G, Wang X, et al. (2012) Overexpression of GOLPH3 promotes proliferation and tumorigenicity in breast cancer via suppression of the FOXO1 transcription factor. Clin Cancer Res 18: 4059-4069.
- Zhang H, Fang J, Yao D, Wu Y, Ip C, et al. (2010) Activation of FOXO1 is critical for the anticancer effect of methylseleninic acid in prostate cancer cells. Prostate 70: 1265-1273.
- Zhang H, Pan Y, Zheng L, Choe C, Lindgren B, et al. (2011) FOXO1 inhibits Runx2 transcriptional activity and prostate cancer cell migration and invasion. Cancer Res 71: 3257-3267.
- Zhao Y, Li X, Cai MY, Ma K, Yang J, et al. (2013) XBP-1u suppresses autophagy by promoting the degradation of FoxO1 in cancer cells. Cell Res 23: 491-507.
- Zhao Y, Li X, Ma K, Yang J, Zhou J, et al. (2013) The axis of MAPK1/3-XBP1u-FOXO1 controls autophagic dynamics in cancer cells. Autophagy 9: 794-796.
- Zhao Y, Tindall DJ, Huang H (2014) Modulation of Androgen Receptor by FOXA1 and FOXO1 Factors in Prostate Cancer. Int J Biol Sci 10: 614-619.
- Bouchard C, Lee S, Paulus-Hock V, Loddenkemper C, Eilers M, et al. (2007) FoxO transcription factors suppress Myc-driven lymphomagenesis via direct activation of Arf. Genes Dev 21: 2775-2787.
- Amin RH, Schlissel MS (2008) Foxo1 directly regulates the transcription of recombination-activating genes during B cell development. Nat Immunol 9: 613-622.
- Mansson R, Welinder E, Åhsberg J, Lin YC, Benner C, et al. (2012) Positive intergenic feedback circuitry, involving EBF1 and FOXO1, orchestrates B-cell fate. Proc Natl Acad Sci U S A 109: 21028-21033.
- Arden KC (2007) FoxOs in tumor suppression and stem cell maintenance. Cell 128: 235-237.
- Chen J, Limon JJ, Blanc C, Peng SL, Fruman DA (2010) Foxo1 regulates marginal zone B-cell development. Eur J Immunol 40: 1890-1896.
- You Y, Zhao H, Wang Y, Carter RH (2009) Cutting edge: Primary and secondary effects of CD19 deficiency on cells of the marginal zone. J Immunol 182: 7343-7347.
- Anzelon AN, Wu H, Rickert RC (2003) Pten inactivation alters peripheral B lymphocyte fate and reconstitutes CD19 function. Nat Immunol 4: 287-294.
- Welinder E, Mansson R, Mercer EM, Bryder D, Sigvardsson M, et al. (2011) The transcription factors E2A and HEB act in concert to induce the expression of FOXO1 in the common lymphoid progenitor. Proc Natl Acad Sci U S A 108: 17402-17407.
- Lin YC, Jhunjhunwala S, Benner C, Heinz S, Welinder E, et al. (2010) A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat Immunol 11: 635-643.
- Timblin GA, Schlissel MS (2013) Ebf1 and c-Myb repress rag transcription downstream of Stat5 during early B cell development. J Immunol 191: 4676-4687.
- Su YW, Hao Z, Hirao A, Yamamoto K, Lin WJ, et al. (2011) 14-3-3sigma regulates B-cell homeostasis through stabilization of FOXO1. Proc Natl Acad Sci U S A 108: 1555-1560.
- Sellars M, Reina-San-Martin B, Kastner P, Chan S (2009) Ikaros controls isotype selection during immunoglobulin class switch recombination. J Exp Med 206:1073-1087.
- Ouyang W, Li MO (2011) Foxo: in command of T lymphocyte homeostasis and tolerance. Trends Immunol 32: 26-33.
- Ouyang W, Liao W, Luo CT, Yin N, Huse M, et al. (2012) Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature 491: 554-559.
- Ouyang W, Beckett O, Flavell RA, Li MO (2009) An essential role of the Forkhead-box transcription factor Foxo1 in control of T cell homeostasis and tolerance. Immunity 30: 358-371.
- Fabre S, Carrette F, Chen J, Lang V, Semichon M, et al. (2008) FOXO1 regulates L-Selectin and a network of human T cell homing molecules downstream of phosphatidylinositol 3-kinase. J Immunol Res. 181: 2980-2989.
- Kerdiles YM, Beisner DR, Tinoco R, Dejean AS, Castrillon DH, et al. (2009) Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat Immunol 10: 176-184.
- Gubbels Bupp MR, Edwards B, Guo C, Wei D, Chen G, et al. (2009) T cells require Foxo1 to populate the peripheral lymphoid organs. Eur J Immunol 39: 2991-2999.
- Kerdiles YM, Stone EL, Beisner DR, McGargill MA, Ch'en IL, et al. (2010) Foxo transcription factors control regulatory T cell development and function. Immunity 33: 890-904.
- Tejera MM, Kim EH, Sullivan JA, Plisch EH, Suresh M (2013) FoxO1 controls effector-to-memory transition and maintenance of functional CD8 T cell memory. J Immunol 191: 187-199.
- Hess Michelini R, Doedens AL, Goldrath AW, Hedrick SM (2013) Differentiation of CD8 memory T cells depends on Foxo1. J Exp Med 210: 1189-1200.
- Anderson G, Jenkinson EJ (2000) Review article: thymus organ cultures and T-cell receptor repertoire development. Immunology 100: 405-410.
- Won HY, Jang EJ, Lee K, Oh S, Kim HK, et al. (2013) Ablation of peroxiredoxin II attenuates experimental colitis by increasing FoxO1-induced Foxp3+ regulatory T cells. Immunol 191: 4029-4037.
- Sykes SM, Lane SW, Bullinger L, Kalaitzidis D, Yusuf R, et al. (2011) AKT/FOXO signaling enforces reversible differentiation blockade in myeloid leukemias. Cell 146:697-708.
- Trinh DL, Scott DW, Morin RD, Mendez-Lago M, An J, et al. (2013) Analysis of FOXO1 mutations in diffuse large B-cell lymphoma. Blood 121: 3666-3674.
- Choi J, Oh S, Lee D, Oh HJ, Park JY, et al. (2009) Mst1-FoxO signaling protects Naïve T lymphocytes from cellular oxidative stress in mice. PLoS One 4: e8011.
Citation: Keniry M, Materon L (2014) Distinctive Roles for the FOXO1 Transcription Factor in Immunological Development, Homeostasis and Malignancy. J Clin Immunol Immunother, 1: 001.
Copyright: © 2014 Megan Keniry, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.