The Autoimmune Diseases Pathogenesis and Prophylaxis
*Corresponding Author(s):
Adams DDFaculty Of Medicine, University Of Otago, Dunedin, New Zealand
Tel:+64 34877989,
Email:duncan.adams@xtra.co.nz
INTRODUCTION
Epidemics of infectious diseases, such as the great plague of London, partly erased by the great fire, were of mysterious origin until Louis Pasteur (Figure 1) introduced the correct Germ Theory of Disease [1], in opposition to the prevailing theory that life was generated spontaneously in decaying organic matter. Lionel Whitby [2] initiated brilliant study of medical bacteriology, which was carried on by others as described by Baron [3], producing amazingly detailed knowledge of Microbiology.

Figure 1: Louis Pasteur, author of The Germ Theory of Disease.
THE IMMUNITY SYSTEM
This exists for defence against infectious diseases, as well described by Arthur Silverstein in William Paul’s classic textbook [4], the description of Nobel Prize highlights summarising major advances well.
AUTOIMMUNE DISEASE
The steps involved in the discovery of the existence and the pathogenesis of the autoimmune diseases are shown in Table 1. As antibodies that fitted specific germs had been discovered, it was natural to think that they were built on a template of antigen. However, Miescher discovered the structure of DNA (Deoxyribonucleic Acid). Then Watson and Crick [5,6] discovered that the order of four bases in a very long double helix provides the genetic code for man and other creatures. Armed with this revolutionary knowledge, Niels Jerne [7] realised that cells are factories that can swiftly convert DNA to RNA to polypeptide, and proposed the Selection Theory, postulating that antibodies are preformed in myriad diversity, awaiting contact with an antigen that fits (Figure 2). Macfarlane Burnet capped this by realizing that it is not antibodies that are selected, but the cells that make them, and that these cells are the lymphocytes, previously thought of as passive spectators of infection (Figure 3). On encountering a complementary antigen, lymphocytes change from having scant cytoplasm to being a blast cell, with copious cytoplasm, able to replicate to form new cells. Burnet realized that the unit of immune specificity is the immunological clone, a subset of lymphocytes with identical receptors for antigen, hence his Clonal Selection Theory of acquired immunity [8].
| Mutations in the V genes of multiplying lymphocytes are the cause of the autoimmune diseases | |
| Haurowitz | Template theory of antibody formation. Antibodies are built on a template of antigen. Wrong. |
| Watson and Crick | Cells are factories able to make myriad different polypeptides by transcribing DNA chains to RNA chains and translating them to polypeptide chains |
| Jerne | The Selection Theory of antibody formation: Antibodies are pre-formed in myriad diversity awaiting selection by an antigen that fits. |
| Burnet [8] | The Clonal Selection Theory: It is not antibodies but their parent lymphocytes that are selected. The unit of immune specificity is the lymphocyte clone, a subset of lymphocytes with identical receptors for antigen. |
| Discovery of autoimmunity | |
| Dameshek [9] | Autoimmune haemolytic anaemia. |
| Freund [10] | Autoimmune destruction of guinea pig testes using adjuvants. |
| Adams and Purves [11] | Thyrotoxicosis |
| Witebsky and Rose [12] | Autoimmune thyroiditis |
| Doniach and Roitt [13] | Thyroid autoimmunity. |
| Porter [14] | Variable region (V) genes code for antigen receptors on lymphocytes and their antibodies. |
| Burnet [15] | The Forbidden Clone Theory: The Forbidden Clones that cause autoimmune disease arise by somatic gene mutations in the V genes of multiplying lymphocytes. |
|
Knight et al., [16] |
Thyroid-Stimulating autoantibodies (TSab) in individual patients contain only one type of light chain, showing origin from mutation in a single lymphocyte |
| Knight and Adams [17] | Absence of allotypic variation in the autoantigen for thyroid stimulating autoantibodies |
| Ben-Nun et al., [18] | Isolation of antigen specific T cell lines capable of mediating autoimmune encephalomyelitis. |
| Cheynier et al., [19] | Somatic mutation occurs in T lymphocyte V genes. |
| Sherwin [20] | Type 1 diabetes is caused by specific destruction of pancreatic islet β cells by cytotoxic T cell forbidden clones |
| Adams [21] | Diabetic retinopathy: Evidence that cytotoxic T cell forbidden clones cause diabetic retinopathy by selectively destroying pericytes. |
Table 1: The Jerne-Burnet Forbidden Clone Theory of Autoimmune Disease.

Figure 2: Niels Jerne, author of the Selection Theory of the Immune Response.

Figure 3: Macfarlane Burnet, author of the Forbidden Clone Theory of Autoimmune Disease.
DISCOVERY OF AUTOIMMUNITY
This happened in 1938 when Dameshek and Schwartz [9], Haematologists, discovered autoimmune haemolytic anaemia. However, this was ignored as an oddity. Then in 1953, Freund et al., [10] produced autoimmune destruction of the testes in guinea pigs by immunising them with testicular tissue and adjuvants that prolonged the antigenic stimulus long enough for the necessary somatic mutations to occur in the responding lymphocytes. Similarly using adjuvants, Witebsky et al., [12] with a JAMA paper, put autoimmunity on the medical map by producing autoimmune thyroiditis in rabbits.
In the same year, Deborah Doniach and Ivan Roitt [13] showed that Hashimoto’s disease of the thyroid is caused by autoimmunity. Then Adams and Purves discovered the Long-Acting Thyroid Stimulator (LATS) that eventually proved to be the thyroid-stimulating autoantibodies that cause thyrotoxicosis [11]. McKenzie [22] confirmed the discovery of LATS, adapting the guinea pig assay to the mouse, which was advantageous.
BURNET’S FORBIDDEN CLONE THEORY
Being a Bacteriologist, used to counting mutation rates in bacteria growing on blood agar plates, Burnet realised that multiplying lymphocytes will mutate similarly, hence his Forbidden Clone Theory of autoimmune disease [15]. This postulates that somatic mutations in the V (Variable region) genes of multiplying lymphocytes produce the aptly named forbidden clones of lymphocytes that cause the autoimmune diseases by reacting with a host antigen instead of a microbial one.
CONFIRMATION OF THE FORBIDDEN CLONE THEORY
This is shown in Table 1, which reports John Knight’s demonstration that Thyroid-Stimulating antibodies (TSab) in individual patients contain only one type of light chain, indicating origin from a single lymphocyte [16]. Additionally, Allison Knight, Cague and Adams set up a precise system for measuring LATS protector [23], which was used to show absence of allotypic variation in the autoantigen for thyroid stimulating autoantibodies [17].
LATS PROTECTOR
Trying to make the LATS assay more specific for small responses, Adams and Kennedy made use of Kriss’ discovery [24] that thyroid extracts will neutralize LATS when incubated with it. However, they found that LATS from different patients showed wide variation in its susceptibility to neutralization. Suspecting an interfering substance, they found that LATS-negative sera from patients with Graves’ disease contained another thyroid autoantibody, which protected LATS from neutralization by competing with it for reaction with the thyroid antigen. Hence, the name LATS protector. The new antibody had no stimulating effect in the mouse bioassay. Baffled, we published this finding [25] and soon had the good fortune to receive the following letter from Deborah Doniach.
A Letter from Deborah Doniach
London, May 5th, 1967.
Dear Duncan,
I wonder why you assume that the new LATS blocking antibody is not active in vivo? It could be more species specific and therefore not show up in the mouse test, yet still have stimulating properties on the human thyroid.
Best wishes,
Deborah Doniach.
LATS PROTECTOR, THE SPECIFIC HUMAN THYROID STIMULATOR
Ivan Roitt was behind this brilliant interpretation, which was entirely logical, as the neutralization was done with human thyroid extracts and the test for activity was performed in the mouse. When mouse thyroid extracts were used, LATS protector disappeared [26]. For final confirmation of the human thyroid-stimulating activity of LATS protector, it was necessary to round up half a dozen courageous Otago Medical School academics prepared to make themselves bioassay animals for LATS protector. This experiment, illustrated in Figure 4, showed that LATS protector does stimulate the human thyroid [27].
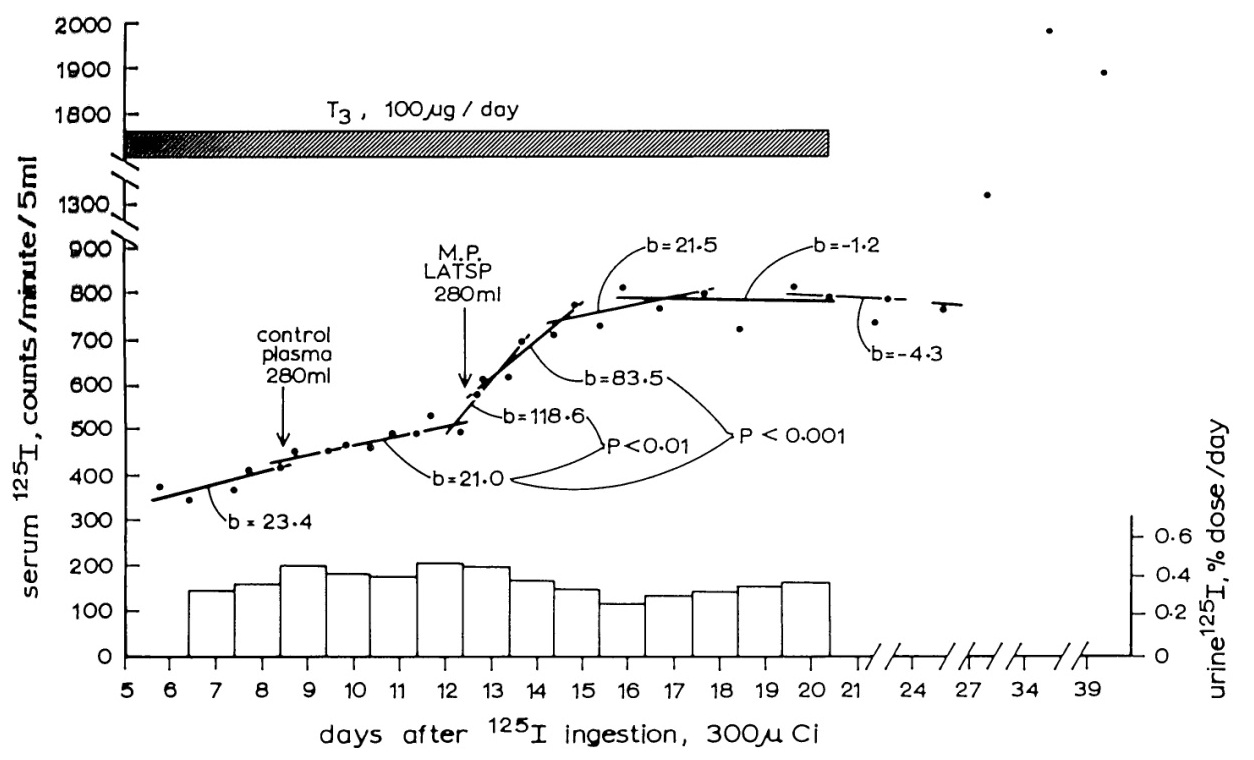
Figure 4: The effect of infusion of LATS Protector (LATSP) into a human volunteer.
A control infusion of normal plasma has had no effect on the slowly rising blood 125I level, but the LATSP infusion has caused a prominent and significant rise, indicating the occurrence of thyroid stimulation [27].
With RDH Stewart and TH Kennedy, Adams showed LATS protector to be present in 90% of cases of untreated Graves’ disease, including all the severe and moderately severe and moderately severe ones. Furthermore, there was a highly significant correlation between a patient’s LATS protector level and her/his thyroid uptake of 131I [28].
Figure 5 illustrates the pathogenesis of Graves’ disease. It originates from a forbidden clone of lymphocytes, which have developed into plasma cells, able to secrete large amounts of Thyroid-Stimulating autoantibodies (TSaab). These stimulate the thyroid gland, causing thyroid hyperplasia and secretion of excessive amounts of thyroid hormone (T4 and T3), which increases the metabolic rate to cause thyrotoxicosis. The high thyroid hormone levels also inhibit the thyrotroph cells of the anterior pituitary gland, by negative feed-back, so TSH secretion ceases, as does response to thyrotrophin-releasing hormone from the hypothalamus.
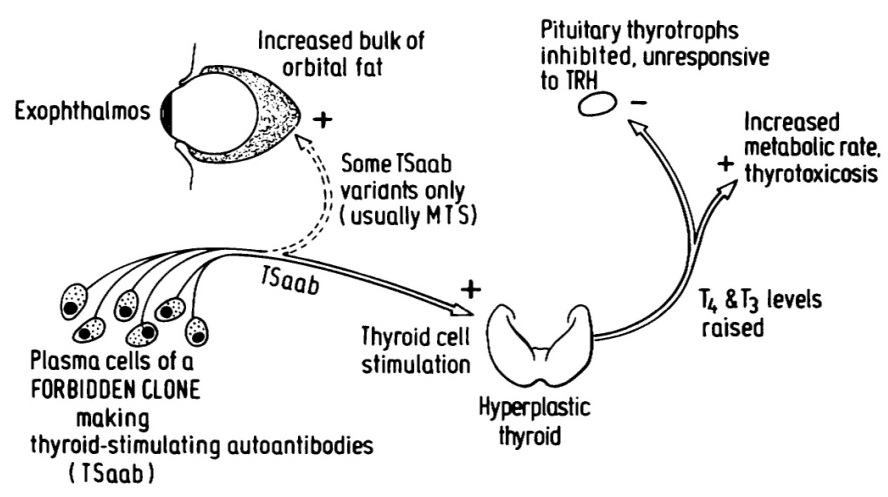
Figure 5: The pathogenesis of Graves’ disease [29].
TSaab from forbidden clones of immunocytes stimulate the thyroid cells, causing overproduction of thyroid hormones T4 and T3 and the manifestations of thyrotoxicosis. The thyrotroph cells of the anterior pituitary are inhibited by the high blood thyroid hormone level, so TSH secretion ceases and response to TRH is absent. Some variants of TSaab react with receptors on fat cells in the orbit to cause exophthalmos from the bulk of adipocyte proliferation, demonstrated by Rundle and Pochin [30].
EXOPHTHALMOS
By ingenious post-mortem measurements, Rundle and Pochin [30] showed that in exophthalmos the component causing the increase in bulk of the extra-ocular orbital contents is fat, indicating that some TSaab variants, usually reactive with the mouse thyroid, cause proliferation of orbital adipose cells. Corticosteroid treatment is needed to inhibit this.
NEONATAL THYROTOXICOSIS
A quintessence of precision in the measurement of a pathogenic autoantibody was achieved by Susan Dirmikis et al., [31] who defined units of LATS protector, then showed that maternal LATS protector levels over 20 units/ml invariably give rise to neonatal thyrotoxicosis in babies, whereas levels below 10 units/ml never do.
GRAVE'S DISEASE A PARADIGM FOR AUTOIMMUNITY
In 1986, at the University of Pisa, Professor Aldo Pinchera led an International Symposium on Thyroid Autoimmunity at which Graves’ disease was presented as a paradigm for autoimmune disease [29].
DISCOVERY OF THE STRUCTURE OF ANTIBODY MOLECULES
Serum, which is blood from which red cells and white cells have been removed, contain dissolved proteins, including the gamma globulins that are now called immunoglobulins because they are the antibodies. How could the atomic structure of such a great mixture of molecules be determined? Henry Kunkel solved this problem by using blood from patients with multiple myeloma, a cancer in which a lymphocyte cell becomes malignant, producing large amounts of a single antibody molecule. Kunkel’s pupil, Gerald Edelman, broke the disulphide bonds holding antibodies together, revealing that each antibody molecule is comprised of four amino acid chains, two long ones and two short ones, the disulphide bonds holding them together to form a functional antibody. Rodney Porter [14] successfully split antibody molecules into their constituent amino acid chains, by using the weak proteolytic enzyme, papain. This produced three fragments, a Crystallizable Fragment, FC, the constant region, and two identical Fragments, FAB, each of which formed identical Antigen Binding sites [32] (Figure 6).
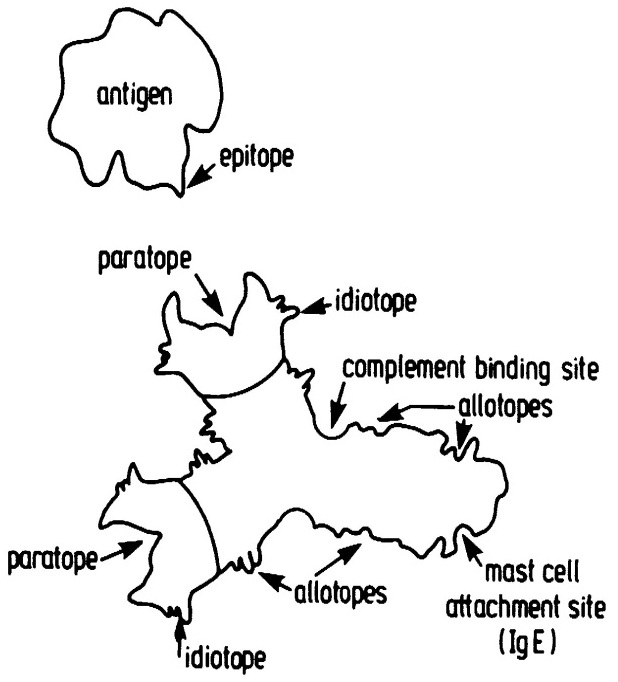
Figure 6: An antibody molecule of IgG class [32]. Showing the two identical paratopes (combining sites for antigen) and the complimentary epitope of the antigen molecule. Also shown are the idiotopes (constant region epitopes) and the attachment site for the C1q molecule of the complement system. Additionally, an attachment site for mast cells present on IgE class antibodies is depicted.
DISCOVERY OF T AND B LYMPHOCYTES
The end of Table 1 reports examples of diseases caused by T cell forbidden clones.
Sherwin’s epic research on Type 1 diabetes [20,34], diabetic retinopathy [21,35], evidence that T lymphocytes undergo somatic mutations similarly to B lymphocytes [19] and Ben-Nun’s classic isolation of antigen-specific T cell lines capable of mediating autoimmune encephalomyelitis [18].
Pathogenic forbidden clones
| Disease or disorder | Autoantigen | Cell type of the forbidden clone |
| Graves’ disease | TSH receptor [29] | B cell, plasma cell |
| Myasthenia Gravis | Acetylcholine receptor [18] | B cell, plasma cell |
| Goodpasture’s disease | On glomerular and lung basement membrane [36] | B cell, plasma cell |
| Pernicious anaemia | Intrinsic factor [37] | B cell, plasma cell |
| Systemic lupus erythaematosis | An intracellular component made copiously available by cytolysis [38] | B cell, plasma cell |
| Thrombosis by lupus anticoagulant [39] | Platelet cell wall phospholipid [40] | B cell, plasma cell |
| Hypocomplementaemia | Alternative pathway C3 convertase “abnormally stabilised by C3 nephritic factor [41] | B cell, plasma cell |
| Haemolytic anaemia | Red cell surface component [42] | B cell, plasma cell |
| Rheumatoid arthritis | Type X1 collagen [43] Calpastatin [44] | B cell, plasma cell |
| Schizophrenia | A neuronal dopaminergic receptor [45,46] | B cell, plasma cell |
| Manic depressive | ?Another neuronal receptor | B cell, plasma cell |
| Systemic scleroderma | ?Fibroblast receptor causing excessive collagen formation [47-49] | B cell, plasma cell |
| Paget’s disease | ?Osteoclast receptor causing bone resorption [50] | B cell, plasma cell |
| Ankylosing spondylitis | Spinal column epithelium component [51] B cell or T cell | B cell, plasma cell |
| Post-streptococcal glomerulonephritis | ?Intracellular glomerular component exposed by nephritogenic strep toxin [52] | ?B cell as Ig is in the lesion |
| Rheumatic fever | Heart component cross-reactive with streptococci [53] | ?T cell as the heart autoantibodies also occur in uncomplicated strep infection [54,55] |
| Diabetes Type 1 | Islet b cell surface component | Cytotoxic T cell, CD8 [20,33] |
| Diabetic retinopathy | On vascular pericyte cells | Cytotoxic T cell, CD8 [34,35] |
| Autoimmune hepatitis? | Hepatocyte surface component | ?Cytotoxic T cell, CD [56] |
| Addison’s disease | ?Adrenocortical cell surface | ?Cytotoxic T cell component [57] |
| Primary biliary cirrhosis | ?Ductal cell surface component [58] | Cytotoxic T cell, CD8 |
| Multiple sclerosis | ?Oligodendrocyte surface component [59] | Cytotoxic T cell, CD8 |
| Polymyositis | ?Muscle cell surface component | CD8 a/b cell [60] |
Table 2: Discovered pathogenic forbidden clones and others awaiting discovery.
THE HISTOCOMPATIBILITY SYSTEM
Discovery by Haldane
This was the discovery of the histocompatibility system, which is essential for defence against virus infection, prevents allo-transplantation [63] and influences the risk of autoimmune disease, as described below.
HUMAN LEUKOCYTE ANTIGENS (HLA)
Functions of the major histocompatibility complex
| 1. The race between virus and cytotoxic T cell. | |||
| The contestants | Replication time | Progeny | |
| Influenza virus | 10 hours [68] | 1,000 virions | |
| Cytotoxic T cell | 18 hours | 2 T cells | |
| The race | Virions | T cells | Virion/T cell ratio |
| Day 1 | 1 | 106 | 1/106 |
| Day 2 | 1 x 1,0002.4 | 106 x 21.3 | 6/1 |
| Day 3 | 2.5 x 1014 | 6.3 x 106 | 107/1 |
| Day 4 | 4 x 1021 | 1.6 x 107 | 1014/1 |
| The result: the virus wins, the patient dies. | |||
| 2. Consequences of the explosive speed of viral replication. Hence, cytotoxic T cell clones need to be 1. Large, preformed (no time for expansion). 2. Specific for conjoint virus-MHC antigenic target, so as not to be muffled by the myriad numbers of free virions. This explains, |
|||
| 1. Why allografts are rejected. | |||
| 2. The strength of allograft rejection, Simonsen phenomenon [69]. | |||
| 3. The need for the MHC restriction phenomenon discovered by Zinkernagel and Doherty [67], the presentation of viral antigens to cytotoxic T cells on host histocompatibility antigens, so that the anti-viral immune attack is directed at the virus-infected cells, the virus factories. | |||
Solution of the genetics of autoimmune disease
THE H GENE THEORY
This example of what Einstein [79] called a “stumble right up against the thing”, enabled Adams and Knight to link the fields of transplantation genetics [63], immunology and autoimmunity, to arrive at the H Gene Theory.
This states that histocompatibility antigen genes, major, minor and HY (the male sex antigen), together with the V (Variable region) genes coding for antigen receptors on B and T lymphocytes, are the germ-line immune response genes, the genes that influence the risk of autoimmune disease [80,81].
MICROBIAL TRIGGERS
Rheumatic carditis and Streptococci: Before the advent of penicillin, rheumatic fever, with crippling or fatal lesions of the heart, was a frequent consequence of infection by β-haemolytic streptococci of Lancefield Group A. This was because of an antigenic similarity between a component of these streptococci and heart tissue, discovered by Kaplan and Meyeserian [83]. Today, with such infection therapeutically aborted by penicillin, rheumatic heart disease, once common, has become rare.
Glomerulonephritis and Streptococci: Post-infective glomerulonephritis follows infection by Group A streptococci of multiple M types. This disease is also less frequent due to use of antibiotics.
Reactive arthritis
Rheumatoid Arthritis (RA) and Proteus mirabilis [84]: Multiple studies over three decades have found high titres of antibodies against this bacterium in a total of 1375 RA patients, but not in other diseases or healthy controls in studies by independent groups in 15 different countries. There was no such elevation in antibodies against 27 other microbial agents. There is evidence that the upper urinary tract is the main site of Proteus infection in RA.
In brilliant studies, inventing diverse biochemical and other technology, Wilson and other members of Ebringer’s team showed shared amino acid sequences between major histocompatibility Class II glycoproteins, type XI collagen and Proteus mirabilis in rheumatoid arthritis [84]. This shows in detail how Proteus mirabilis triggers the autoimmune attack on the joint collagen of patients with HLA-DR1/4.
Ankylosing Spondylitis (AS) and Klebsiella: In world-wide studies involving 1330 AS patients and 1191 healthy controls, the AS patients showed significantly increased antibody titres to Klebsiella. There is evidence that the gut is the main site of Klebsiella infection in AS.
Schizophrenia and virus infection: Acute schizophrenia has been observed to follow upper respiratory tract virus infection and John Knight has assembled much evidence that schizophrenia is an autoimmune disease caused by autoantibodies that react with neuronal receptors influencing the limbic system of the brain [47,86].
Recently, Fabienne Brilot [87], a Paediatrician, studying post-infectious encephalomyelitis in children, suspecting an autoimmune basis, developed a new method of detecting antibodies to cell surface dopamine-2 receptors. It would be of great interest to test blood from acute schizophrenics with this method, to see if it contains the autoantibodies that cause schizophrenia.
Seeking antigenic triggers and their corresponding autoantigen, with excellent technology at the NIH, Laing et al., [88] immunized rabbits with neurotropic strains of influenza virus, inducing autoantibodies to a brain-specific 37-kDa protein. This needs further exploration, as does the whole field of psychotic aetiology, until the presumptive forbidden clones have been demonstrated with the clarity obtained for Graves’ diseases and myasthenia gravis.
INFORMATION FROM SEQUENCING ANTIGENS ON TRIGGERING BACTERIA
Ebringer’s Basic Books: Details of the development of the methods used for successful determination of the amino acid sequences of antigens on the rheumatoid arthritis-triggering bacteria, Proteus mirabilis are described in the book, “Rheumatoid arthritis and Proteus” by Ebringer [89] Figure 7. Similarly, the book “Ankylosing spondylitis and Klebsiella”, also by Ebringer [90], describes how the amino acid sequences of antigens on Klebsiella pneumoniae, that enormously increase the risk of ankylosing spondylitis, were determined, and how they exert their effect.
Figure 7: Professor Alan Ebringer, BSc, MD, FRCP, FRACP.
Discovered that Proteus mirabilis in the upper urinary tract triggers rheumatoid arthritis.
Discovered that Klebsiella pneumoniae in the gut triggers ankylosing spondylitis.
Confirmed the H Gene Theory by finding two antigens on Proteus, one resembling HLA-DR1/4 the predisposing HLA antigen, one resembling the autoantigen attacked.
Showed how HLA-B27 predisposes, 69-fold, to ankylosing spondylitis.
Confirmation of the H Gene theory: This research provides experimental confirmation, at the molecular level, of the H Gene Theory of the inheritance of the autoimmune diseases, described above, in confirming the speculated presence of multiple antigens on triggering bacteria and alternative clonal development enabling development of the forbidden clones that cause the associated autoimmune diseases.
How HLA-DR1/4 predisposes to rheumatoid arthritis [44]: Figure 8 shows space-filling models of the amino acid sequences of the histocompatibility antigen HLA-DR1/4 and the Proteus mirabilis haemolysin antigen. Close structural similarity is apparent. This means the immune tolerance imposed by the histocompatibility antigen will extend to this Proteus antigen, preventing immune reaction with it.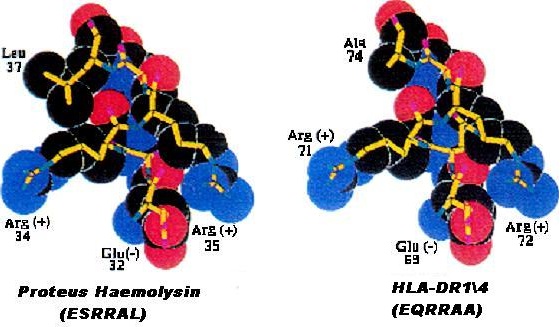
Figure 8: Molecular similarity between histocompatibility antigen HLA-DR1/4 and Proteus haemolysin, preventing immune reaction against this bacterial antigen [89].
Figure 9 shows space-filling models of the amino acid sequences of the Proteus mirabilis urease antigen and Type 11 collagen, an autoantigen attacked in rheumatoid arthritis. The urease antigen is completely different from HLA-DR1-4, so will not be protected from immune reaction, being free to stimulate development of a forbidden clone reacting with the closely similar Type 11 collagen molecule, an autoantigen attacked in rheumatoid arthritis.
Figure 9: Molecular similarity of Proteus urease and Type XI collagen an autoantigen attacked in rheumatoid arthritis. This explains how infection with Proteus mirabilis causes rheumatoid arthritis [89].
How HLA-B27 predisposes to ankylosing spondylitis [44]: Figure 10 shows space-filling models of the amino acid sequences of the histocompatibility antigen HLA-B27 and two antigenic peptides on the bacterium Klebsiella pneumoniae. The Klebsiella nitrogenase antigen closely resembles HLA-B27, so will be covered by the tolerance induced by HLA-B27, but the bacterium’s pullanase peptide is different and able to stimulate development of a forbidden clone attacking the spine to cause ankylosing spondylitis.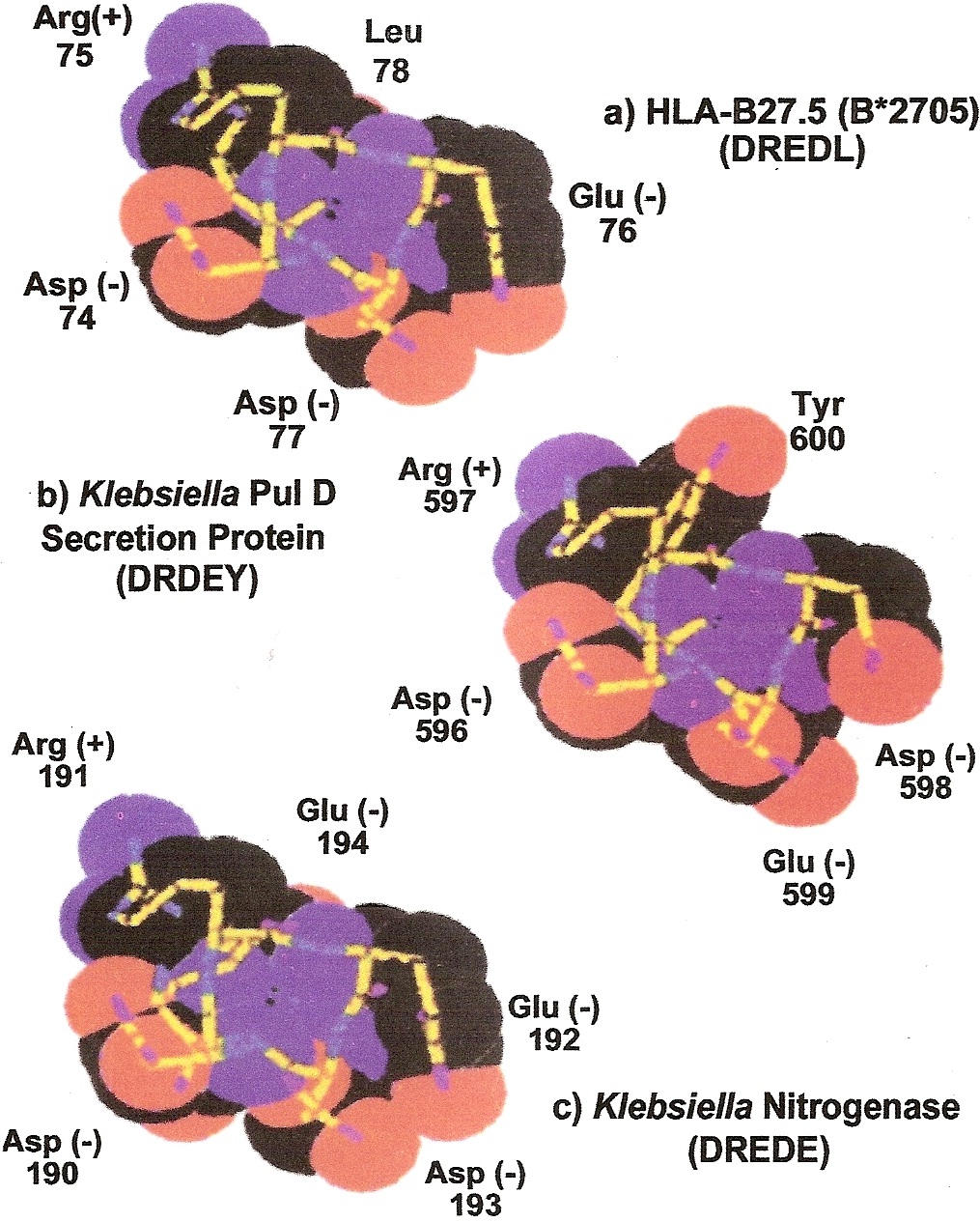
Figure 10: Molecular similarity of HLA-B27 and the Klebsiella nitrogenase bacterial antigen, preventing immune reactivity, but dissimilarity with the Klebsiella pullonase bacterial antigen, allowing immune reactivity which can lead to development of the forbidden clones that cause ankylosing spondylitis, explaining the strong genetic predisposition (69-fold) by HLA-B27 discovered by Terasaki. (Adams DD, Knight JG, Ebringer A (2010) Autoimmune diseases. Autoimmunity Reviews 9: 525-530).
PROPHYLAXIS OF AUTOIMMUNE DISEASES
Paralysis a rare autoimmune complication of universal virus infection?
The Salk (killed) and Sabin (attenuated) polio vaccines have been brilliantly successful in preventing the polio leg paralyses. This exemplifies how autoimmune diseases in general, can be prevented by finding and vaccinating against their microbial triggers.
The lead in prophylaxis given by the polio vaccines: The Salk (killed) and Sabin (attenuated) polio vaccines have been brilliantly successful in preventing the polio leg paralyses. This exemplifies how autoimmune diseases in general, can be prevented by finding and vaccinating against their microbial triggers.
Finding microbial triggers: Ebringer has succeeded in this with rheumatoid arthritis and ankylosing spondylitis. He has pioneered this new field of medical research, developing a whole new technology that needs to be copied in other diseases, especially schizophrenia. Systematic studies of autoimmune diseases, with collaboration between clinicians and microbiologists are needed.
Prevention of ankylosing spondylitis: There is 69-fold increased risk of ankylosing spondylitis in males with HLA-B27 [71]. Ebringer’s discovery that the bacterium Klebsiella pneumoniae triggers ankylosing spondylitis offers prevention of this disease by vaccination of HLA-B27 positive boys with Klebsiella pneumoniae. This could initiate a new form of prophylaxis, which could culminate in prevention of schizophrenia, once its triggering microbe (probably viral) has been identified.
DISCUSSION
Some disease associations are cross-tissue autoimmunity, for example the eye proptosis of Graves’ disease, caused by variants of the thyroid-stimulating autoantibodies that react with receptors on orbital fat cells [30], and diabetic retinopathy [21], probably caused by destruction of retinal pericyte cells by antigenic variants of the T cell forbidden clones that destroy the pancreatic islet β cells to cause Type 1 diabetes.
Many autoimmune diseases, such as Graves’ disease, already have satisfactory therapy. Immunotherapy, by radiological or chemical immune ablation, with immune reconstitution by autologous bone marrow cells, pioneered by Tyndall [91], can be used to save the lives of patients with dangerous autoimmune diseases, such as systemic scleroderma [92].
Selective destruction of forbidden clones could be achieved by isolating their autoantigen (such as the TSH receptor of Graves’ disease, cloned by Vassart and Dumont) [93] and attaching it to a cytotoxic moiety, such as bungarotoxin or 131I iodine (emitting short-range β particles), then administering the molecular complex intravenously to destroy the pathogenic clones of plasma cells. When monoclonal antibody technology was discovered, it was mistakenly assumed that this would provide cures for the autoimmune diseases, a notion greatly encouraged by the drug companies. Struggling to help his patients get benefit from use of Rituximab, Dreyfus [94] envisages major progress from anti-viral therapies and ultimately virus vaccines.
Prevention is better than cure, so finding and countering antigenic triggers of autoimmune diseases is the ideal. Recognition of the universality of microbial triggers of autoimmune diseases is a major advance, in showing how the diseases can be prevented by finding and vaccinating against their triggers. Ebringer has led the way by discovering three triggers and developing the technology for finding others.
REFERENCES
- Dubois RJ (1951) Louis Pasteur. Gollancz, London.
- Whitbury L, Hynes M (1953) Medical Bacteriology. Churchill, London.
- Baron S (1996) Medical Microbiology. (4th edn), University of Texas, Galveston, USA.
- Paul WE (1999) Fundamental Immunology. (4th edn), Lippincott-Raven, Philadelphia, USA.
- Watson JD, Crick FH (1953) Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 171: 737-738.
- Watson JD, Crick FH (1953) Genetical implications of the structure of deoxyribonucleic acid. Nature 171: 964-967.
- Jerne NK (1955) The natural-selection theory of antibody formation. Proc Natl Acad Sci USA 41: 849-857.
- Burnet FM (1959) The clonal selection theory of acquired immunity. Cambridge University Press, London.
- Dameshek W, Schwartz SO (1938) Haemolysins as cause of chemical and experimental haemolytic anaemias, with particular reference to nature of spherocytosis and increased fragility. Amer J Med Sci 196: 769.
- Freund J, Lipton MM, Thompson GE (1953) Aspermatogenesis in the guinea pig induced by testicular tissue and adjuvants. J Exp Med 97: 711-726.
- Adams DD, Purves HD (1956) Abnormal responses in the assay of thyrotrophin. Proc Univ Otago Med School 34: 11-12.
- Witebsky E, Rose N, Terplan K, Paine JR, Egan RW (1957) Chronic thyroiditis and autoimmunisation. JAMA 164: 1439-1447.
- Doniach D, Roitt IM (1957) Auto-immunity in Hashimoto’s disease and its implications. J Clin Endocrinol Metab 17: 1293-1304.
- Porter RR (1959) The hydrolysis of rabbit y-globulin and antibodies with crystalline papain. Biochem J 73: 119-126.
- Burnet M (1959) Auto-immune disease. I. Modern immunological concepts. Br Med J 2: 645-650.
- Knight J, Laing P, Knight A, Adams D, Ling N (1986) Thyroid-stimulating autoantibodies in individual patients contain only one type of light chain, showing origin from mutation in a single lymphocyte. J Clin Endocrinol Metab 62: 342-347.
- Knight A, Adams DD (1983) Absence of allotypic variation in the autoantigen for thyroid stimulating autoantibodies. Clin Exp Immunol 52: 317-324.
- Ben-Nun A, Wekerle H, Cohen IR (1981) The rapid isolation of clonable antigen-specific T lymphocyte lines capable of mediating autoimmune encephalomyelitis. Eur J Immunol 11: 195-199.
- Cheynier R, Henrichwark S, Wain-Hobson S (1998) Somatic hypermutation of the T cell receptor V beta gene in microdissected splenic white pulps from HIV-1-positive patients. Eur J Immunol 28: 1604-1610.
- Sherwin RS (2000) Diabetes mellitus. In: Goldman L, Bennett JC (eds.). Text book of Medicine. (21st edn). Saunders, Philadelphia. Pg: 1893-1898.
- Adams DD (2008) Autoimmune destruction of pericytes as the cause of diabetic retinopathy. Clin Ophthalmol 2: 295-298.
- McKenzie JM (1958) Delayed thyroid response to serum from thyrotoxic patients. Endocrinology 62: 865-868.
- Knight A, Cague WS, Adams DD (1980) Measurements of the thyroid-stimulating autoantibodies. In: Rose NR, Friedman H (eds.). Manual of Clinical Immunology. (2nd edn). American Society for Microbiology, Washington. Pg: 391-402.
- Kriss JP, Pleshakov V, Chien JR (1964) Isolation and identification of the long-acting thyroid stimulator and its relation to hyperthyroidism and circumscribed pretibial myxedema. J Clin Endocrinol Metab 24: 1005-1028.
- Adams DD, Kennedy TH (1967) Occurrence in thyrotoxicosis of a gamma globulin which protects LATS from neutralization by an extract of thyroid gland. J Clin Endocrinol Metab 27: 173-177.
- Adams DD, Kennedy TH (1971) Evidence to suggest that LATS protector stimulates the human thyroid gland. J Clin Endocrinol Metab 33: 47-51.
- Adams DD, Fastier FN, Howie JB, Kennedy TH, Kilpatrick JA, et al. (1974) Stimulation of the human thyroid by infusions of plasma containing LATS protector. J Clin Endocrinol Metab 39: 826-832.
- Adams DD, Kennedy TH, Stewart RD (1974) Correlation between long-acting thyroid stimulator protector level and thyroid 131I uptake in thyrotoxicosis. Br Med J 2: 199-201.
- Adams DD, Knight A, Knight JG, Laing P (1987) Graves’ disease: a paradigm for autoimmunity. In: Pinchera A, Ingbar SH, McKenzie JM, Fenzi GF (eds.). Thyroid Autoimmunity. Plenum, New York. Pg: 1-10.
- Rundle F, Pochin E (1944) The orbital tissues in thyrotoxicosis: a quantitative analysis relating to exophthalmos. Clin Sci 5: 51-74.
- Munro DS, Dirmikis SM, Humphries H, Smith T, Broadhead GD (1978) The role of thyroid stimulating immunoglobulins of Graves's disease in neonatal thyrotoxicosis. Br J Obstet Gynaecol 85: 837-843.
- Adams DD (1983) Autoimmune Mechanisms. In: TF Davies (ed.). Autoimmune Endocrine Disease. John Wiley & Sons, New York. Pg: 1-39.
- Mitchison NA (1954) Passive transfer of transplantation immunity. Proc R Soc Lond B Biol Sci 142: 72-87.
- Sutherland DE, Goetz FC, Sibley RK (1989) Recurrence of disease in pancreas transplants. Diabetes 1: 85-87.
- Klintworth GK (1988) The eye. In: Rubin E, Farber JL (eds.). Pathology. (2nd edn). Lippincott, Philadelphia. Pg: 1456-1483.
- Lindstrom J, Shelton D, Fujii Y (1988) Myasthenia gravis. Adv Immunol 42: 233-284.
- Saus J, Wieslander CM, Langerveld JPM, Qinones S, Hudson BG (1988) Identification of the Goodpasture antigen as the alpha 3 (IV) chain of collagen IV. J Biol Chem 1988; 263: 13374-13380.
- Roitt IM, Doniach D, Shapland C (1965) Autoimmune phenomena in relation to gastric mucosa in human disease. In: Grabar P, Miescher PA (eds.). Immunopathology. IV International Symposium. Schwabe, Basel. Pg: 314-324.
- Adams DD (1982) Systemic lupus erythaematosus: a simple concept of the pathogenesis and its genetic basis. In: Dawkins RL, Christiansen FT, Zilko PJ (eds.). Immunogenetics in rheumatology. Amsterdam: Excerpta Medica. Pg: 242-243.
- Exner T (1989) Lupus anticoagulant. Today’s Life Sciences. 1: 40-46.
- Boey ML, Colaco CB, Gharavi AE, Elkon KB, Loizou S, et al. (1983) Thrombosis in systemic lupus erythematosus: striking association with the presence of circulating lupus anticoagulant. Br Med J (Clin Res Ed) 287: 1021-1023.
- Davis AE 3rd, Ziegler JB, Gelfand EW, Rosen FS, Alper CA (1977) Heterogeneity of nephritic factor and its identification as an immunoglobulin. Proc Natl Acad Sci USA 74: 3980-3983.
- Kaplan ME (1985) Autoimmune haemolytic disease due to warm-reacting antibodies. In: Wyngaarden JB, Smith LM (eds.). Textbook of Medicine. (17th edn), Saunders, Philadelphia. Pg: 908-910.
- Wilson C, Ebringer A, Ahmadi K, Wrigglesworth J, Tiwana H, et al. (1995) Shared amino acid sequences between major histocompatibility complex Class II glycoproteins, type XI collagen and proteus mirabilis in rheumatoid arthritis. Ann Rheum Dis 54: 216-220.
- Ménard HA, el-Amine M (1996) The calpain-calpastatin system in rheumatoid arthritis. Immunol Today 17: 545-547.
- Knight JG (1982) Dopamine-receptor-stimulating autoantibodies: a possible cause of schizophrenia. Lancet 2: 1073-1076.
- Knight JG, Knight A, Pert CB (1987) Is schizophrenia a virally-triggered anti-receptor autoimmune disease? In: Helmchen H, Henn FA (eds.). Biological perspectives of schizophrenia. Wiley, New York. Pg: 107-127.
- Potter SR, Bienenstock J, Goldstein S, Buchanan WW (1985) Fibroblast growth factors in scleroderma. J Rheumatol 12: 1129-1135.
- Xu WD, Leroy EC, Smith EA (1991) Fibronectin release by systemic sclerosis and normal dermal fibroblasts in response to TGF-beta. J Rheumatol 18: 241-246.
- Gilliland BC (2001) Systemic sclerosis. In: Braunwald E, Fauci AS, Kasper DL, Hauser S, Longo D, et al. (eds.). Harrison’s Principles of Internal Medicine. (15th edn), McGraw-Hill, New York, USA. Pg: 1937-1946.
- Fallon MD, Schwamm HA (1989) Paget’s disease of bone. An update on the pathogenesis, pathophysiology, and treatment of osteitis deformans. Pathol Annu 24 Pt 1: 115-159.
- Ebringer A (2013) Ankylosing spondylitis and Klebsiella. Springer, London.
- Williams DG, Peters DK (1987) Glomerulonephritis and renal manifestations of systemic disease. In: Weatheral DJ, Ledingham JGG, Warrell DA (eds.). Oxford Textbook of Medicine. (2nd edn), University Press, Oxford, USA. Pg: 36-55.
- Bisno AL (1985) Rheumatic fever. In: Wyngaarden JB, Smith LM (eds.). Textbook of Medicine. (17th edn), Saunders, Philadelphia. Pg: 1527-1533.
- Tagg JR, McGiven AR, Guthrie DA (1972) Heart-reactive antibodies in rheumatic fever. Med J Aust 1: 621-624.
- Dienstag JL, Isselbacher KJ (2001) Autoimmune hepatitis. In: Braunwald E, Fauci AS, Kasper DL, Hauser SL, Longo DL, et al. (eds.). Harrison’s Principles of Internal Medicine. (15th edn), McGraw-Hill, New York, USA. Pg: 1750-1752.
- Williams GH, Dluhy RG (2001) Disorders of the adrenal cortex. In: Braunwald E, Fauci AS, Kasper DL, Hauser SL, Longo DL, et al. (eds.). Harrison’s Principles of Internal Medicine. (15th edn), McGraw-Hill, New York, USA. Pg: 2084-2105.
- Rubin E, Faber J (1994) Primary biliary cirrhosis. In: Rubin E, Farber JL (eds.). Pathology. (2nd edn), Lippincott, Philadelphia. Pg: 740-743.
- Hauser SL, Goodkin GE (2001) Multiple sclerosis and other demyelinating diseases. In: Braunwald E, Fauci AS, Kasper DL, Hauser SL, Longo DL, et al. (eds.). Harrison’s Principles of Internal Medicine. (15th ed), McGraw-Hill, New York, USA. Pg: 2452-2461.
- Banker BQ, Engel AG (1986) The polymyositis and dermatomyositis syndromes. In: Engel AC, Banker BQ (eds.). Myology: basic and clinical. McGraw Hill, New York, USA. Pg: 1385-1422.
- Klein J (1975) Biology of the mouse histocompatibility-2 complex. Berlin, Springer.
- Haldane JBS (1933) The genetics of cancer. Nature 132: 266-267.
- Auchinloss H, Sykes M, Sacks DH (1999) Transplant Immunology. In: Fundamental Immunology. (4th edn), Lippincott-Raven, Philadelphia, USA.
- Dausset J, Svejgaard A (1977) HLA and disease. Munksgaard, Copenhagen, USA.
- McKusick VA (1978) Mendelian inheritance in man. Catalogs of autosomal dominant, autosomal recessive and X-linked phenotypes (5th edn). Johns Hopkins University Press, Baltimore, USA.
- Adams DD (1987) Protection from autoimmune disease as the third function of the major histocompatibility gene complex. Lancet 2: 245-249.
- Zinkernagel RM, Doherty PC (1974) Restriction of in vitro T cell-mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature 248: 701-702.
- Fenner F, White DO (1975) Medical Virology. Academic Press, London, UK. Pg: 75.
- Simonsen M (1969) On the nature and measurement of antigenic strength. Transplant Rev 3: 22-35.
- Vladutiu AO, Rose NR (1971) Autoimmune murine thyroiditis relation to Histocompatibility (H-2) type. Science 174: 1137-1139.
- Schlosstein L, Terasaki PI, Bluestone R, Pearson CM (1973) High association of an HL-A antigen, W27, with ankylosing spondylitis. N Engl J Med 288: 704-706.
- Tiwari JL, Terasaki PI (1985) HLA and disease associations. Springer, New York, USA.
- Bielschowsky M, Helyer BJ, Howie JB (1959) Spontaneous haemolytic anaemia in mice of the NZB/BL strain. Proc Univ Otago Med Sch 37: 9-11.
- Howie JB, Helyer BJ (1968) The immunology and pathology of NZB mice. Adv Immunol 9: 215-266.
- Knight JG, Adams DD (1978) Three genes for lupus nephritis in NZB x NZW mice. J Exp Med 147: 1653-1660.
- Drake CG, Babcock SK, Palmer E, Kotzin BL (1994) Genetic analysis of the NZB contribution to lupus-like autoimmune disease in (NZB x NZW) F1 mice. Proc Natl Acad Sci USA 91: 4062-4066.
- Kono DH, Burlingame RW, Owen DG, et al. (1994) Lupus susceptibility loci in New Zealand mice. Proc Natl Acad Sci USA 91: 10,168-10,172.
- Knight JG, Adams DD (1981) Genes determining autoimmune disease in New Zealand mice. J Clin Lab Immunol 5: 165-170.
- Einstein A (1934) Essays in science. Philosophical Library Inc, New York. Pg: 92.
- Adams DD, Knight JG (1980) H gene theory of inherited autoimmune disease. Lancet 1: 396-398.
- Knight JG, Adams DD (1982) The genetic basis of autoimmune disease. Ciba Found Symp : 35-56.
- Shevack EM (1998) Organ-specific autoimmunity. In: WE Paul (ed). Fundamental Immunology. (4th edn), Lippincott-Raven, New York. Pg: 1089-1125.
- Kaplan MH, Meyeserian M (1962) An immunological cross-reaction between group-A streptococcal cells and human heart tissue. Lancet 1: 706-710.
- Rashid T, Ebringer A (2007) Rheumatoid arthritis is linked to Proteus--the evidence. Clin Rheumatol 26: 1036-1043.
- Ebringer A, Rashid T, Wilson C, Ptaszynska T, Fielder M (2006) Ankylosing spondylitis, HLA-B27, and Klebsiella pneumoniae - an overview proposed for early diagnosis and treatment. Curr Rheumatol Rev 2: 55-68.
- Knight JG (1982) Dopamine-receptor-stimulating autoantibodies: a possible cause of schizophrenia. Lancet 2: 1073-1076.
- Dale RC, Merheb V, Pillai S, Wang D, Cantrill L, et al. (2012) Antibodies to surface dopamine-2 receptor in autoimmune movement and psychiatric disorders. Brain 135: 3453-3468.
- Laing P, Knight JG, Hill JM, Harris AG, Oxford JS, et al. (1989) Influenza viruses induce autoantibodies to a brain-specific 37-kDa protein in rabbit. Proc Natl Acad Sci U S A 86: 1998-2002.
- Ebringer A (2012) Rheumatoid arthritis and Proteus. Springer, London.
- Ebringer A (2013) Ankylosing Spondylitis and Klebsiella. Springer, London.
- Farge D, Passweg J, van Laar JM, Marjanovic Z, Besenthal C, et al. (2004) Autologous stem cell transplantation in the treatment of systemic sclerosis: report from the EBMT/EULAR Registry. Ann Rheum Dis 63: 974-981.
- Englert H, Katelaris C, McGill N, Schrieber L, Moore J (2005) 'Grape-sultana' sign represents a favourable response to aggressive treatment of early diffuse systemic scleroderma. Intern Med J 35: 436-437.
- Costagliola S, Morgenthaler NG, Hoermann R, Badenhoop K, Struck J, et al. (1999) Second generation assay for thyrotropin receptor antibodies has superior diagnostic sensitivity for Graves’ disease. J Clin Endocrinol Metab 84: 90-97.
- Dreyfus DH (2011) Autoimmune disease: A role for new anti-viral therapies? Autoimmun Rev 11: 88-97.
Citation: Wesolowski R, Carson III WE (2015) Tumor Infiltrating Lymphocytes-The Next Step in Assessing Outcome and Response to Treatment in Patients with Breast Cancer. J Clin Immunol Immunother 2: 005.
Copyright: © 2015 Adams DD, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.