Introduction
The liver is the largest metabolically active exocrine gland of the body and produces bile that serves as the excretory route for endogenous and exogenous compounds. Bile also assists in digestion and absorption of fat by providing bile acids and phospholipids to the duodenum and plays an immunological role by delivering IgA to the intestine. The inability of the liver to produce bile is termed cholestasis, which accompanies many liver diseases and can be caused by endogenous as well as exogenous compounds. Agents that increase bile formation are known as choleretic agents, while agents that decrease bile formation are known as cholestatic agents. Bile formation is an energy-dependent process and requires vectorial transport of solutes from the sinusoidal space to the canaliculus. The coordinated function of transporters located at the sinusoidal and canalicular membranes results in the accumulation of solutes (primarily organic anions including bile salts and glutathione) in the canalicular space providing the osmotic driving force for bile formation [1-7]. It is thus easy to appreciate the paradigm that cholestasis results when the ability of the liver to transport solutes into the canaliculus is compromised. In cholestatic liver diseases [2,8], compounds normally excreted in the bile accumulate, due to impairment of hepatic transport functions, in the liver and blood resulting in adverse effects. Our knowledge of mechanisms underlying bile formation and cholestasis has been steadily increasing. The aim of this review is to summarize our current understanding of the role of p38 MAPK and its isoforms in bile formation and cholestasis.
Transporters in bile formation
Several transporters are involved in hepatocellular transport of organic as well as inorganic solutes [5]. Three of these transporters, namely Na+-Taurocholate Cotransporting Polypeptide (NTCP), Bile Salt Export Pump (BSEP) and Multidrug Resistance Protein 2 (MRP2) are primarily involved in vectorial transport of organic anions involved in bile formation. NTCP mediates Na+/TC cotransport across the sinusoidal membrane. BSEP and MRP2 are ATP-Binding Cassette (ABC) transporters, located at the canalicular membrane and mediate canalicular secretion of bile salts and conjugated organic anions, respectively [9-12]. The coordinated function of NTCP and BSEP allows transport of conjugated bile salts into the canalicular space providing the stimulus for that fraction of bile known as bile-dependent bile formation. It should be noted that while some bile salts (taurocholate, TC and tauroursodeoxycholate, TUDC) produce choleresis, others (taurolithocholate, TLC and taurodeoxycholate, TDC) are known to produce cholestasis [2,13]. The bile-acid independent fraction of bile results from biliary excretion of other solutes including conjugated organic anions transported by MRP2 [14]. Transporter regulation: It is now well established that apart from genetic defects [15-18], regulation of these transporters at the level of transcriptional, translational and post-translational modifications may be altered by chemicals/disease processes. Indeed, cholestasis is associated with down-regulation of NTCP and MRP2 [19,20] with a relatively preserved expression of BSEP [19]. It is becoming evident that transcriptional regulation of hepatocellular transporters is primarily mediated via the nuclear receptor superfamily [3,21], while the post-translational regulation is mediated via classical second messengers. The transcriptional changes are delayed effects of cholestasis that assure long term adjustments of transporter functions. On the other hand, post-translational regulations involve short-term rapid changes in Plasma Membrane (PM) localization of transporters [1,5,6] and these permit rapid changes in bile formation. This review focusses on short-term regulations. The long term transcriptional regulations have recently been reviewed by others [3,21]. Short-term regulation: A transporter must be translocated to the appropriate membrane for it to transport its solute across that membrane. This is a complex regulated process requiring participations of various signaling molecules along with vesicles and cytoskeletons. A breakdown in this regulated process can lead to a decreased amount or an absence of a transport protein at its intended site resulting in decreased or no transport function and hence cholestasis. It is becoming evident that a translocation defect is an important event in cholestasis. Thus, experimental cholestasis induced by bile duct ligation, endotoxin, TLC and Estradiol-17beta-D-glucuronide (E-17G) is associated with decreased level of NTCP [22], MRP2 [20] and BSEP [23,24] in the plasma membrane. Certain other agents (cAMP, TC, TUDC) have been shown to stimulate translocation of these transporters to the membrane [12,25-29]. Acute cholestasis induced by TLC and E-17G is associated with the retrieval of BSEP and MRP2 from the canalicular membrane and these effects are reversed by cAMP [23,24,30]. TUDC has been shown to reverse TLC-induced retrieval of MRP2 [27,31]. These results highlight the importance of translocation defect in acute cholestasis. Thus, an understanding of the cellular mechanisms regulating transporter translocation and retrieval is necessary for medical management of patients with cholestasis. A number of signaling pathways including Protein Kinase Cs (PKCs), p38 Mitogen Activated Protein Kinases (p38MAPKs), Rab proteins, Protein Phosphatases (PPs), actin binding proteins, ca2+ and Phosphoinosotide-3-Kinases (PI3Ks), [1,5,6,32,33] have been implicated in the regulation of PM localization of transporters (NTCP, BSEP and MRP2) by cAMP, TUDC, E17G and TLC [1,5,34,35]. In this review, we discuss the regulation by p38 MAPK as the role of p38 MAPK has not recently been reviewed.
P38 MAPK
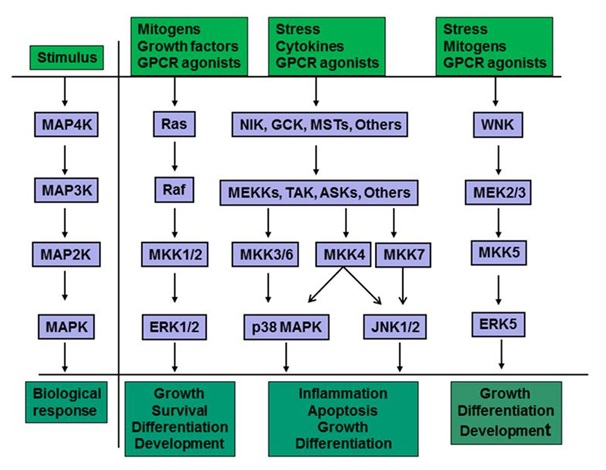
Mammalian cells have four major types of MAPKs cascade and these include ERK1/2, JNKs, p38 MAPK and ERK5 cascades [36-40] (Figure 1). Each of these cascades consists of a core module of three tiers of protein kinases termed MAPK, MAP2K, and MAP3K. There are seven MAP2Ks (also known as MEK, MAP/ERK kinase, or MKK) that differentially activate different MAPKs by dual phosphorylation on Thr and Tyr. Thus, ERK1/2 is activated by MKK1/2, p38 MAPKs are activated by MKK3, MKK4, and MKK6, JNKs by MKK4/7 and ERK5 by MKK5. The MAPKs regulate members of a family of protein Ser/Thr kinases termed MAPK-Activated Protein Kinases (MAPKAPKs). The MAPKAPKs are related enzymes that respond to extracellular stimulation through direct MAPK-dependent activation loop phosphorylation and kinase activation [42]. The deactivation of MAPKs is achieved through dephosphorylation catalyzed by MAPK-specific Phosphatases (MKPs) including dual specific MAPK phosphatases [43,44]. MAPKs mediate/regulate diverse cellular functions including embryogenesis, apoptosis, immunity, proliferation, and differentiation by integrating signals from intra- and extracellular stimuli [41,45-47]. The p38 MAP kinase pathway shares many similarities with the other MAP kinase cascades, being associated with inflammation, cell growth, cell differentiation and cell death [48,49].Activation stimuli: p38 MAPK is also known as Stress-Activated Protein Kinase (SAPK) since it is mostly activated by stresses such as ER stress, oxidative stress, metabolic stress and DNA damage, and by inflammatory cytokines [42,50-54]. The activation of p38 MAPK by a variety of stimuli (Figure 1) involves dual phosphorylation on Thr180 and Tyr182 by upstream MAPK kinases. While three upstream MAPK Kinases (MKKs), namely MKK3, MKK6 and MKK4, control the activation of p38 MAPK, MKK3 and MKK6 are considered the major activators of p38 MAPK [55-58]. A MAPKK-independent mechanism of p38 activation involves TAB1 (Transforming growth factor-beta-Activated protein Kinase 1 (TAK1)-binding protein 1) [59]. Interacting of TAB1 with p38 MAPK induces p38 autophosphorylation [60]. Inactivation of p38 MAPK involves dephosphorylation by phosphatases targeting threonine and tyrosine residues in the activation loop [61]. Phosphatases involved are Protein Phosphatase (PP) 2A add PP2C or Striatal Enriched Tyrosine Phosphatase (STEP) and Haemopoietic Tyrosine Phosphatase (HePTP) [49]. Inhibition of PP2A increases p38 activity by various stimuli [62-64]. In addition, microRNAs contribute to the homeostatic signaling of p38 MAPK pathway [40]. P38 MAPK isoforms: There are four known isoforms (α, β, γ and δ) of p38 MAPK with 60% homology and only α and β isoforms are expressed in livers [49,65,66]. Whereas p38α is ubiquitously expressed, p38β is mostly found in the brain, p38γ is predominantly expressed in skeletal muscle and p38δ gene expression is found in the lung, kidney, testis, pancreas and small intestine [49,67]. Activation of isoforms by upstream kinases differs based on stimuli and cell type [49,68,69]. In general, MKK6 can phosphorylate all four p38 MAPK family members, where as MKK3 activates p38α, p38γ and p38δ, but not p38β. In non-hepatic cells, MKK6 preferentially activates p38β MAPK [70] and MKK3 activates only p38α and p38 MAPKs [71]. Thus, MKK3 is expected to activate p38α and not p38β MAPK in the liver. In addition, p38α can also be phosphorylated by MKK4. Cyclic AMP has been shown to specifically activate p38α, but not p38β in adipocytes [72]. Selective activation of p38 isoforms has been reported in other cells [47,73,74]. Studies also suggest isoform specific effects of p38 MAPK. For example, p38α MAPK is involved in inflammation induced by proinflammatory cytokines, IL-1 and TNFα [75]. P38α and β MAPK have been shown to mediate cell cycle [67]. P38α but not p38β MAPK regulates chronic pain development [76]. P38α promotes apoptosis and p38β inhibits apoptosis [77,78]. P38δ synergistically regulates ERK pathway, whereas p38α medicates nuclear factor kappa B (NFкB) pathway for drug resistance [79].Hepatic effects of p38 MAPK: Many hepatic functions are affected by p38MAPK. These functions include regulation of proliferation [80], protection against hypoxic injury [81], gluconeogenesis [82], enhancement hepatic ketogenesis [83], improvement of hepatic steatosis [84], inhibition on the progression of liver fibrosis [85], involvement in the pathogenesis and progression of intrahepatic cholestasis of pregnancy [86], bile acid synthesis [87] and anti-apoptotic effect of TUDC [88], bile acid-induced apoptosis [89,90], and biliary excretion of bile acids [91]. The p38 MAPK also regulates transporter translocation. For example, p38 MAPK is involved in GLUT4 translocation [92], EGF receptor endocytosis [93] and serotonin transporter trafficking [94]. In the liver, the p38 MAPK pathway is involved in the PM translocation/retrieval of MRP2 and BSEP [95,96]. Note that PM translocation of NTCP is not regulated by p38 MAPK [97,98]. Thus, the regulation of PM translocation/retrieval of MRP2 and BSEP by p38 MAPK in hepatocytes is discussed below.
Hepatocellular Transporter Translocation and P38 MAPK
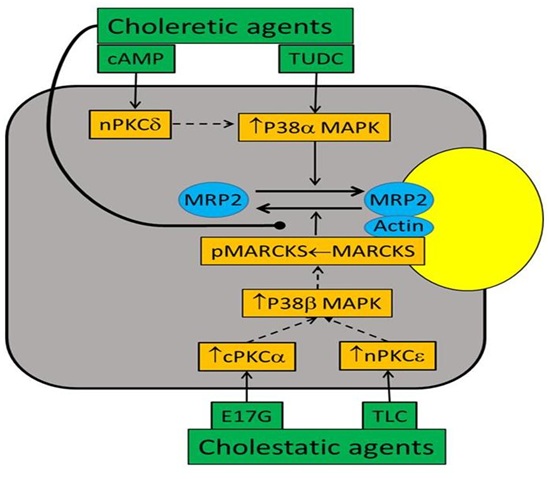
Both choleretic (cAMP, TUDC, TC) and cholestatic agents (TLC, TDC and E17-G) have been employed to determine the cellular mechanisms regulating PM localization of BSEP and MRP2. These studies suggest a role for p38 MAPK in PM insertion/retrieval of BSEP and MRP2. TUDC-induced increases in bile acid secretion and BSEP translocation to the canalicular membrane require PI3K-independent activation of p38 MAPK [91,96]. Translocation of MRP2 to PM by cAMP is also mediated by p38 MAPK [99]. E-17G-induced endocytosis of BSEP and MRP2 is associated with decreased bile flow and biliary excretion of BSEP/MRP2 substrates and p38 inhibition prevents the internalization of BSEP/MRP2 [95]. P38 MAPK plays a role in oxidative stress-induced retrieval of BSEP and MRP2 [100]. TLC activates p38 MAPK [101] and decreases PM localization of MRP2 in hepatocytes [27,102]. These results suggest that p38 MAPK is involved in the insertion of BSEP/MRP2 to PM by choleretic agents as well as the retrieval of BSEP/MRP2 from PM by cholestatic agents. These paradoxical effects of p38 MAPK on retrieval as well as insertion of BSEP/MRP2 to PM led to the hypothesis that the effects of p38 MAPK on insertion and retrieval of these transporters may be mediated via different p38 MAPK isoforms. This hypothesis is based on findings that p38α MAPK and p38β MAPK have reciprocal effects on the expression of inducible nitric oxide synthase in renal mesangial cells [103].There are limited studies on the role of p38 MAPK isoforms in the retrieval/insertion of hepatic transporters. One study showed that cAMP selectively activates p38α via activation of MKK3 and knockdown of p38α MAPK inhibited cAMP-induced insertion of MRP2 in a hepatic cell line [99]. These results were confirmed in studies with hepatocytes from MKK3 knockout mice [104]. In contrast to cAMP, TLC did not activate MKK3 or p38α MAPK in wild type mice hepatocytes and still decreased PM-MRP2 in MKK3 knockout hepatocytes. Additionally, TLC activated MKK6 in MKK3 knockout hepatocytes, and knockdown of p38β MAPK abrogated TLC-mediated decreases in PM-MRP2 in a hepatic cell line [104]. Taken together, these results suggest that activation of the MKK3/p38α MAPK pathway facilitates plasma membrane insertion of MRP2, whereas activation of the MKK6/p38β MAPK pathway mediates retrieval of PM-MRP2 by TLC (Figure 2). Whether insertion/retrieval of BSEP is also similarly regulated by P38 MAPK isoforms remains to be established.
Crosstalk between P38 MAPK and PKC Signaling Pathways
Cellular mechanisms by which activation of p38 MAPK leads to alteration of PM localization of MRP2 may involve interactions with other signaling pathways. There are studies suggesting that Protein Kinase C (PKC) isoform also differentially regulate PM localization of BSEP and MRP2. PKC comprises a family of at least 12 isoforms [105], which include conventional (cPKCα, β, βI, βII and ), novel (nPKCδ, ε, and ), atypical (aPKC and ) isoforms and PKC. cPKCα, nPKCδ, nPKCε, aPKC and probably cPKCβII are present in hepatocytes [106-108]. For example, cPKCα mediates MRP2 and BSEP retrieval by E17G [109] and PMA [110], nPKCδ mediates cAMP-induced translocation of MRP2 to the plasma membrane [111], and nPKCε is responsible for the internalization of MRP2 by TLC [102]. Since PKCs activate p38MAPK in hepatocytes [95,112-114], it is likely that PKCs act upstream of p38 MAPK in hepatocytes (the regulation of PM localization of BSEP and MRP2). Indeed, results of several studies are consistent with this hypothesis. For example, hypoxia induced activation of p38 MAPK is blocked by PKC inhibitors in chicken hepatocytes [112]. Activation of p38 MAPK by PKCδ and PKCε has been reported in ischemic preconditioning in rat hepatocytes [115]. nPKCδ has been suggested to initiate p38 MAPK activation in butyrate-induced apoptosis in human colon adenoma cells [116]. One study suggests that cPKCs act upstream of p38 MAPK in E17G-induced retrieval of PM-BSEP and PM-MRP2 [95]. These studies raise the possibility that different PKCs may activate different p38MAPKs and this may explain isoform-specific effects of PKCs and p38MAPK on PM-MRP2. More specifically, activation of p38α MAPK by nPKCδ may facilitate MRP2 translocation to PM and activation of p38β MAPK by cPKCα and/or nPKCε may promote retrieval from PM (Figure 2). A preliminary study (unpublished) by authors showing that cAMP fails to activate p38α MAPK in PKCδ knockout hepatocytes is consistent with this hypothesis. However, further studies will be required to define whether PM-MRP2 localization involves isoform-specific effects of PKCs on p38α and p38β MAPKs in hepatocytes.
Liver Functions, Inflammatory Diseases and P38 MAPK
The possibility of p38 MAPK as the therapeutic target for Rheumatoid Arthritis (RA) was raised with the finding in 1994 that a p38 inhibitor blocked Lipopolysaccharide (LPS)-induced TNFα and IL1β production by monocytes [75]. P38 MAPK inhibitor appeared to be a potential “wonder drug” and work began in earnest to synthesize and clinically evaluate novel inhibitors for inflammatory diseases [117-119]. These compounds were mainly competitive antagonists that blocked ATP binding to the kinase [120]. AP-1-dependent gene expression is p38 isoform specific in human breast cancer cells [121]. However, potency, lack of selectivity and toxicity limited their utility [122]. These compounds inhibited p38α and β but not the γ or δ isoforms [123]; at higher concentrations many other kinases were blocked [124]. While effective in preclinical models, a variety of toxicity problems, especially affecting the liver, interfered with clinical development [117]. Clinical trials with certain p38 MAPK inhibitors were discontinued because of liver toxicity [117,125], which is likely to be due to the inhibition of p38α MAPK. This is consistent with studies suggesting that deficiency of p38α in the liver increases the expression of chemokines to recruit more inflammatory cells [126], facilitates N-Nitrosodiethylamine (DEN) - induced hepatocellular carcinoma and increased proliferation [127,128]. In addition, p38α MAPK facilitates bile formation by translocating MRP2 to the PM. Recent studies suggest that MKK6 is a potential therapeutic target in RA [120,129]. Since MKK6 activates p38β but not p38α MAPK in hepatocytes and p38β MAPK induces cholestasis by retrieving MRP2 from the PM, MKK6 inhibitors may be less toxic to the liver. A better understanding of underlying signaling pathways of p38 MAPK should allow us to develop the drug target limiting hepatic toxicity and thereby improving the efficacy of p38 MAPK inhibitors in inflammatory diseases.In summary, our understanding of cellular mechanisms underlying bile formation and cholestasis is steadily increasing. Recent studies suggest that p38α MAPK facilitates bile formation by inserting MRP2 into PM, while p38β MAPK mediates cholestasis by retrieving MRP2 from the PM. It appears that isoform specific effects of PKCs may be mediated via p38 MAPK isoforms. Since p38 MAPK is also involved in inflammation, development of drugs for inflammatory diseases by inhibiting p38 MAPK should take into account the effect of p38 MAPK isoforms in bile formation and cholestasis.
Acknowledgement
The authors thank Dr. Webster for helpful discussion and Trena Haroutunian for secretarial assistance. The studies by authors referenced here were in part supported by a National Institutes of Health grant DK-33436.