ABSTRACT
What is already known on this topic?
Glycerol Kinase Deficiency (GKD) [OMIM 307030] is a rare X-linked recessive metabolic disorder, which is characterized biochemically by hyperglycerolaemia and glyceroluria. Mutations in the GK gene are the major causes of isolate GKD.
What this study adds?
We report a novel splice-site mutation (c.1340 -2A>G) identified at GK gene analysis.
Glycerol Kinase Deficiency (GKD) is a rare X-linked recessive metabolic disorder. Mutations in the GK gene are the major causes of isolate GKD. In this study, we report an isolated GKD patient, who exhibited elevated serum triglycerides and urine glycerol. Mutational analysis of GK gene using Sanger sequencing revealed a novel splice-site mutation (c.1340 -2A>G), and sequence analysis of the RT-PCR product confirmed that this mutation resulted in a skip of exon17. The mRNA of GK in patient was decreased by 59.2%, compared to the control. This finding expands the mutation spectrum of GK, helps us understand the molecular basis of GKD, also contributes to contemporary genetic counseling.
KEYWORDS
Glycerol kinase deficiency; Glycerol kinase gene; Hyperglycerolemia; Splice-site mutation
Introduction
Glycerol Kinase Deficiency (GKD) [OMIM 307030] is a rare X-linked recessive metabolic disorder, which is characterized biochemically by hyperglycerolemia and glyceroluria [1]. The clinical spectrum varies from asymptomatic and Central Nervous System (CNS) deterioration to severe developmental delay, and is found to be related with GK gene mutation. The gene (GK) is located on Xp21 and encodes a key enzyme in regulation of glycerol uptake and metabolism that catalyzes the phosphorylation of glycerol [2].
GKD can be categorized into three types: Infantile GKD, Juvenile GKD and Adult GKD. The Infantile GKD, also called the complex GKD or Xp21 contiguous gene deletion syndrome, is the most common type of GK mutation, which is caused by a deletion of GK gene or the neighboring genes, the clinical manifestations depends on the deletion of the genes [3]. The juvenile GKD is called symptomatic GKD, with symptoms like episodic vomiting, acidemia and Central Nervous System (CNS) deterioration. The adult GKD is called asymptomatic GKD, which detected incidentally with pseudohypertriglyceridemia. The juvenile and adult GKD are also called isolated GKD, which mainly caused by point mutations in GK gene.
In this case report, a Chinese patient clinically diagnosed with isolated glycerol kinase deficiency is detected to have a novel splice-site mutation in the GK gene, which leads to a skip of exon 17.
Case Report
General information and clinical manifestations
A 3-month-old male term infant was referred to hospital due to no weight gain for three months. He was born with uterine-incision delivery route due to malpresentation after a first pregnancy of a 27-year-old mother at 38 weeks of uncomplicated gestation. His Apgar score was 9 points at 5min, with birth weight 3250g and head circumference 32cm. The parents were nonconsanguineous.
Laboratory tests
The infant’s serum triglyceride was 11.17mmol/l (normal range: 0.52-1.56mmol/l), and his urine analysis revealed increased urine glycerol. His serum cortisol level was 1.00μg/dl (normal range: 3.7-19.4μg/dl (8 am); 2.9-17.3μg/dl (4 pm)), testosterone was 1.02ng/ml (normal range:1.42-9.23ng/ml), lactate dehydrogenase was 275.5U/l (normal range: 109-245U/l), and creatine kinase was 77.0U/l (normal range: 24-190U/l).
Molecular studies
The clinical and laboratory findings were consistent with the clinical diagnosis of isolated glycerol kinase deficiency. The proband and his mother were recruited at the Hunan Jiahui genetics hospital. The study was approved by the Ethics Committee of the State Key Laboratory of Medical Genetics (SKLMG) and an informed consent was signed by the patient’s parents before testing.
Mutation screening of the GK gene by Sanger sequencing:
Genomic DNA was extracted from peripheral blood lymphocytes, using the Quick gene DNA Whole Blood Kit L (FUJIFILM, Tokyo, Japan). All 19 exons and flanking regions of the GK gene (NM_000167) were amplified by Polymerase Chain Reaction (PCR) using the primers. The primer sequences and PCR conditions are available upon request. The PCR products were confirmed by Polyacrylamide Gel Electrophoresis (PAGE), followed by Sanger sequencing (Biosune, Changsha, China). The sequencing results were analyzed using the DNASTAR (Madison, WI, USA) software.
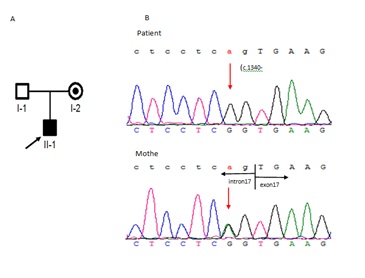
We identified a split-site mutation (c.1340 -2A>G) in the GK gene (NM_000167), his mother was heterozygous (Figure 1) for the mutation, and it had not been previously reported in the Human Gene Mutation Database (HGMD), dbSNP144, or Exome Variant Server (EVS). To investigate the effect of this splice-site mutation on RNA species, we performed Reverse transcription-PCR on RNA from the patient’s peripheral blood samples.
Analysis of GK expression by real-time quantitative PCR:
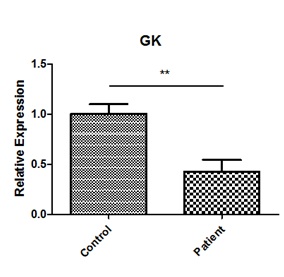
Total RNA was extracted from the patient’s immortalized lymphoblastusing Trizol reagent in accordance with standard procedures, first strand cDNA synthesis, was carried out as previously described. Quantitative PCR was performed on a real-time PCR system (step one plus, ABI, USA) using a Eva Green 2X qPCR Master Mix -ROX (abm) following the cycling conditions: 95°C for 10min followed by 40 cycles of 95°C for 10s, and 60°C for 1min. The relative standard curve method was used to analyze the expression level. The expression was normalized to Actin in the same sample and three biological repeats were measured. The real-time primer pairs for GK were 5′-GGTAGATGGAGGAATGACCAGCAAC-3′ and 5′-GTTTCGGGCATTGAGGGCTT-3′ (product length: 98bp); and real-time primer pairs for Actin were 5′-CGTCTTCCCCTCCATCGT-3′ and 5′-GAAGGTGTGGTGCCAGATTT-3′ (product length: 184bp). Real-time PCR showed that GK mRNA in patient was decreased by 59.2%, compared to their age- and gender-matched controls (Figure 4).
Discussion
Currently, there are about 34 mutations in GK gene (NM_000167) have been reported in patients with GKD (HGMD Professional 2017.1), including missense/nonsense mutations (60-61%), splice-site mutations (21-22%), deletion mutations(15-16%) and insertion mutations (3-4%).
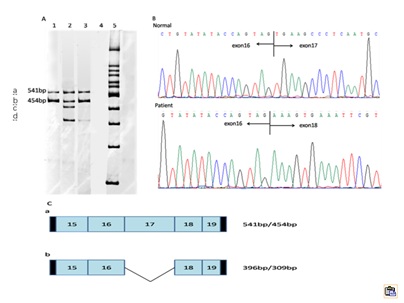
The GK gene (NM_000167) contains 19 exons and yield 1575bp transcript. We identified a patient with isolated GKD due to splice-site mutation (c.1340 -2 A>G). RT-PCR analysis of the patient’ s peripheral blood lymphocyte cell revealed that there were four fragments, two wild-type GK mRNA, and two mutant mRNA. The mutation created a cryptic splice acceptor site, resulting in alternative splicing of the transcript with a small amount of correctly spliced GK mRNA. Likewise, we found that there were normal splicing in the splice-site mutations reported previous [4,5]. Sequencing the fragments, we found that the mutation caused a skipping of the whole exon17, leading a frame shift mutant of p. Val446Glufs*48. Then, we did the real-time PCR, and found that the GK mRNA in patient was decreased by 59.2%, compared to the control. So, we can get a conclusion that the splice-site mutation (c.1340 -2 A>G) of GK may be the cause of the patient.
Previous studies had also reported the deletion of exon17 in two isolated GKD patients, one presented psychomotor, growth retardation and multiple fractures from minor trauma, which couldn’t be prevented by vitamin D or phosphate supplements, and another one manifested pale, somnolent, hyperventilate and severe abdominal pains [6,7]. It’s suggested that the deletion of exon17 is strongly associated with the incidence of GKD.
Interestingly, most of the patients with split-site mutation reported previously presented with asymptomatic form of GKD [4,5,7-9]. Only one patient from the literature (IVS9-1G>T) who had a history of metabolic and CNS crises at the age 6 years and 3 months, indicating the symptomatic form of GKD, but he was uneventful in the neonatal period and early childhood [10].
Previous studies showed that there was no genotype-phenotype correlation in GKD [1,11,12]. Our patient presented no symptoms, excepting no weight gain. The probable causes may be as followings: First, it may be related to age, just like the patient reported by Hellerud et al. [10], the patient didn’t present symptoms at an early age. Second, the patient may still have part of normal mRNA, so the clinical symptom is mild. Third, it may be related to Nonsense Mediated Decay (NMD). Zhang et al., found that aberrant mRNA generated by splice site mutations of the GK gene can be eliminated by NMD in the asymptomatic form of GKD [9]. In contrary, there was no NMD in our study, and reviewed the other split-site mutations previously reported, no NMD was found too [4,5]. Fourth, some studies demonstrated that glycerolutilization by the cells (especially, hepatocytes) is rate-limited by the membrane permeation step, rather than the GK step [13-15]. AQP9, an aquaglyceroporin, has been reported to be of great significance in glycerol membrane permeation step [13-15]. Expression of AQP9 is influenced by gender and disease state. In human, despite exhibiting similar hepatic AQP9 protein, women manifest lower hepatocyte glycerol permeability compared to men [16]. In obese subjects with Non-Alcoholic Fatty Liver Disease (NAFLD), the hepatic expression of AQP9 was decreased [16]. In the rodent, female rats have lower levels of hepatic AQP9 protein compared with males in both fed and fasting conditions. The mice model of NAFLD, demonstrated decreased hepatocyte AQP9 and glycerol permeability correlated with increased plasma glycerol [16], andAQP9-/- mice exhibited a marked increase in plasma glycerol and triglyceride levels compared with AQP9+/- mice, revealing a deficient glycerol metabolism [17]. Therefore, in addition to the GK mutation that causes a high serum triglyceride, the possible role of AQP9 needs to be emphasized, but this still needs to be further studied in GKD patients.
Glycerol kinase deficiency has a suspected risk for pancreatitis and coronary heart disease, and there is no effective way to cure GKD, but there may be some treatment, such as aggressive Therapeutic Lifestyle and combination lipid-altering drug, to reduce the serum glycerol.
Conclusion
We identified a novel splice-site mutation c.1340 -2 A>G in the GK gene, which was most likely responsible for this patient’s pathogenic cause. This detection offered an important insight into the pathogenic mutation spectrum of glycerol kinase deficiency and could help in clinical diagnosis and genetic counseling of patients with glycerol kinase deficiency.
Conflict of Interest
There is no conflict of interest.
Acknowledgment
We thank the patient’s family for their help and support. This study was supported by the National Natural Science Foundation of China (Grant number 81501268, 81571450) and the Key Research and Development Program of Hunan Province (Grant number 2016WK2029).
Figures
Figure 1: The pedigree chromatogram of the GKD family. The arrow indicates the proband. I-2 is a carrier. B: c.1340 -2 A>G sequencing results of the patient and his mother.
Figure 2: A: Lane1 - normal control (two transcripts, 541bp and 454bp). Lane2 - patient. Lane3 -patient’s mother. Lane4 - blank. Lane 5 - Marker (100bp ladder), we named the four products a, b, c, and d. B: Direct sequencing of the RT-PCR products across the site of the c.1340 -2 A>G mutation, which revealed a skip of the exon17. C: Normal splicing and aberrant splicing produced 541bp/454bp and 396bp/309bp PCR fragments, respectively, the black boxes represent the primers.
Figure 3: (1) Sequencing results blast of the products a and b, query is a, and the other is b; (2) Sequencing results blast of the products a and c, the above one is a, the other is c; (3) Sequencing results blast of the products b and d, the above is b, the below is d.
Figure 4: GK and actin mRNA expression in patient and control. Patient and control total RNA were screened for GK and actin mRNA transcripts by real-time RT-PCR. GK mRNA in patient was decreased by 59.2%, compared to their age- and gender-matched controls.
Tables
| Samples | Mn(theo)a × 10-4 | Mn(NMR)b ×10-4 | Mn(GPC)c ×10-4 | Mw/Mnc | Rhd (nm) | ?-potential (mV) |
| EM27 | 0.78 | 0.83 | 0.82 | 1.03 | 4.5 | 18.2 |
| EM53 | 1.36 | 1.41 | 1.11 | 1.02 | 4.3 | 24.2 |
| EM106 | 2.52 | 2.58 | 1.51 | 1.02 | 6.1 | 25.4 |
| AEA | 3.21 | 3.26 | 2.32 | 1.42 | 6.1 | -14.4 |
Table 1: Number-average Molecular weight (Mn), Molecular weight distribution (Mw/Mn), hydrodynamic radius (Rh), and ?-potential for the polymers.
aCalculated from Equation (2), bEstimated from 1H NMR, cEstimated from GPC, dEstimated from DLS.
| PIC micelles | Mwa × 10-5 | Rga | Rhb | Rg/Rh | Naggc | dPICd | ?-potential (mV) |
| (nm) | (nm) |
| AEA/EM27 | 8.48 | 15.1 | 15.2 | 0.99 | 50 | 0.096 | -0.88 |
| AEA/EM53 | 189 | 36.6 | 41.0 | 0.89 | 735 | 0.109 | -0.53 |
| AEA/EM106 | 111 | 28.6 | 32.4 | 0.88 | 302 | 0.129 | -0.20 |
Table 2: Dynamic and static light scattering data for PIC micelles in 0.1 M NaCl.
aEstimated by SLS in 0.1 M NaCl, bEstimated by DLS in 0.1 M NaCl, cAggregation number of PIC micelles calculated from Mw(SLS) of PIC micelles determined by SLS and Mw of the corresponding unimers determined by 1H NR and GPC, dDensity calculated from Equation (3).