Severe Acute Hyponatremia as an Initial Presentation of Acute Intermittent Porphyria Triggered by a Subdermal Etonogestrel Implant
*Corresponding Author(s):
Jean WheelerDepartment Of Anesthesia, Critical Care & Emergency Medicine, Emory University School Of Medicine, 660 Ralph McGill Blvd NE, Atlanta, GA 30312, Atlanta, United States
Fax:+1 4047785194
Email:jwheel9@emory.edu
Abstract
Case description: A 19-year-old African-American female with history of childbirth two months prior presented to the emergency department of Grady Memorial Hospital with vague but severe back pain and found with marked hyponatremia of 125mEq/L. Serum sodium level decreased to 113mEq/L after volume resuscitation with 0.9% sodium chloride. The patient experienced progressive decline in mental status and a single generalized tonic clonic seizure. Patient was admitted to intensive care unit and improved with administration of 3% sodium chloride. Extensive evaluation for etiology of euvolemic hyponatremia was initially unremarkable, and patient was managed with free water restriction, hypertonic sodium chloride, vasopressin receptor antagonists, and antihypertensive medications. Empiric removal of a recently inserted etonogestrel implant was performed with resolution of patient’s symptoms. Approximately 2 weeks following hospital discharge, the send-out lab for urine porphobilinogen was found to be notably elevated.
Conclusion: We stress the importance of considering the diagnosis of AIP in patients presenting with back or abdominal pain, hyponatremia and altered mental status who are reproductive age females and using implanted hormonal contraceptive devices. Appropriate supportive treatment and removal of the implant is required to prevent morbidity and life-threatening consequences.
Keywords
INTRODUCTION
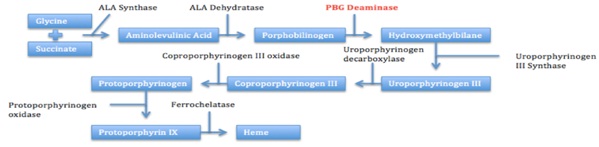
CASE DESCRIPTION
She recovered well and elected to have an etonogestrel 68 mg implant (Implanon?, Merck & Co., Inc., Whitehouse Station, NJ, USA) placed in her left arm for contraception, which was performed two weeks prior to this presentation. The patient continued to produce and pump breast milk throughout this time. The day prior to admission, she presented to an Emergency Department (ED) at another hospital for mid-back pain and fatigue, was diagnosed with a viral syndrome, and discharged home. Notable laboratory studies from that visit revealed sodium of 131mEq/L and creatinine of 1.26mg/dL (baseline 0.7-0.8). The following morning the patient continued to experience severe back pain. The patient’s parents noticed worsened fatigue, prompting presentation to our ED for further evaluation. On the day of admission, the patient also described three days of decreased appetite and nausea.
Initial physical examination was unremarkable; the patient’s back was not tender to palpation, nor could pain be elicited by musculoskeletal or costovertebral angle maneuvers. Laboratory evaluation revealed a sodium level of 125mEq/L and a creatinine of 1.1mg/dL. Initial imaging, including CT renal stone protocol and CT Head without contrast, was unrevealing. The ED physicians assessed the patient as having hypovolemic hyponatremia, and administered a two liter bolus of 0.9% sodium chloride (NaCl). Following this, the patient’s mental status deteriorated and repeats sodium was decreased to 116mEq/L, prompting consultation to the Medical Intensive Care Unit (MICU) service for progressively symptomatic and worsening acute hyponatremia. On MICU evaluation, repeat physical examination suggested patient was currently at or near euvolemia. On neurological exam, she was now obtunded and unresponsive to verbal or painful stimuli with a disconjugate gaze and absent patellar reflexes. Sinus tachycardia to 120s and marked hypertension with systolic blood pressures in the 200s were noted. She was admitted to the MICU for intensive monitoring of sodium and acute treatment of hyponatremia and hypertension. While en route to ICU, she suffered a witnessed general tonic clonic seizure of two minutes duration prompting emergent intubation for airway protection.
Immediate laboratory data post-seizure revealed sodium level of 113mEq/L. Urine sodium of 174mEq/L, urine osmolality of 473mOsm/kg, and serum osmolality of 245mOsm/kg again suggesting euvolemic hyponatremia and consistent with elevated ADH state. Fluid restriction and infusion of 3% NaCl at 30mL/hr were started. As malignant hypertension (possibly from post-partum eclampsia) leading to hypertensive encephalopathy and seizure could not be immediately excluded, a nicardipine infusion was initiated for acute blood pressure control. Over the next 24 hours, sodium level increased to 124mEq/L, and infusion of 3% hypertonic saline was decreased to 15mL/hr. No further seizure activity occurred and acute encephalopathy and focal neurologic symptoms improved steadily, facilitating liberation from mechanical ventilation within 24 hours. Table 1 demonstrates further diagnostic evaluation undertaken to determine etiology of apparent SIADH state on admission [6,7]. While the underlying etiology of patient’s elevated ADH state was unknown initially, it is important to note that several of patient’s symptoms, including pain and nausea, are likely to have contributed to her elevated ADH state.
| Etiologies of SIADH | Patient findings/results |
|
Other causes of excessive ADH Hypothyroidism Adrenal insufficiency |
Normal TSH and cosyntropin stimulation test |
|
Malignancy Lung |
Negative chest, abdomen, and pelvis CT scans; negative transvaginal ultrasound |
|
Infection Tuberculosis |
Negative chest CT, absence of clinical symptoms consistent with infection Negative HIV testing during pregnancy per OSH records and Negative HIV testing during current hospitalization |
|
Intracranial processes Brain tumors |
Negative non contrast head CT and brain/spine MRI Normal hypothalamic-pituitary-ovarian axis labs, current lactation |
|
Medications/Ingestions SSRIs |
Was not on any of these medications at presentation Drug/volatile screen negative, with exception of elevated acetone level from starvation ketosis |
|
Others |
Elevated urine porphobilinogen - result available after hospital discharge Psychiatric consultation did not suggest postpartum psychosis |
After consultation with nephrology, conivaptan was started, along with continuation of 3% hypertonic saline, and led to improved serum sodium and osmolality. As all other evaluations were negative on admission, AIP moved increasingly higher on the differential diagnosis for this patient. As such, OB-GYN was consulted for removal of the etonogestrel implant, which occurred on day 4 of hospitalization. Nicardipine infusion was stopped and transitioned to oral amlodipine and hydralazine. The patient was then transferred to the floor. On hospital day 6, hypertonic saline was held, followed by slow weaning of conivaptan, with sodium levels remaining between 132 to 139mEq/L. With resolution of hyponatremia, her back pain and hypertension also improved, permitting discontinuation of opiate pain medications and oral anti-hypertensives; however, she continued to have persistent, asymptomatic tachycardia. The patient’s appetite returned, with unusual cravings for high carbohydrate containing foods. She was discharged on hospital day 13 and was seen in clinic two weeks later with a sodium level of 138 and resolved tachycardia.
Approximately 5 days after hospital discharge, the physician was notified of an elevated urine porphobilinogen (a send-out lab that normally takes 2-3 weeks to result) of 56.80mg/g creatinine (Reference Range < 2.0mg/g creatinine). Patient was informed of this finding and follow up with Benign Hematology Clinic was arranged and encouraged for further evaluation including genetic analysis. Despite repeated contact with patient via telephone and encouragement to return for outpatient evaluation, patient refused further assistance and has since been lost to follow-up.
DISCUSSION
Six weeks after a recent childbirth, our patient elected to have an etonogestrel implant inserted into her arm as a contraceptive method. Two weeks later, the patient experienced symptoms and laboratory findings consistent with AIP. In review of porphyrinogenic medications listed on the Porphyria Foundation’s drug database, progestins are listed as unsafe due to their ability to induce accumulation of porphyrins [16]. We believe that the progestin-only contraception, in addition to her recent decreased caloric intake, triggered this AIP crisis and, ultimately, established the patient as suffering from porphyria. Following removal of etonogestrel implant and discontinuation of other porphyrinogenic medications (such as nicardipine), the patient’s clinical status, including symptomatic hyponatremia, dramatically improved. Despite studies demonstrating the association of AIP attacks with oral contraception [17,18], importantly and to our knowledge, none have reported the role of an implanted contraceptive in precipitating overt AIP. Adding to the unusual nature of our patient’s case is the rarity of AIP among African and African American populations. A review published in 2000 noted that only 38 patients have been reported with AIP and African ancestry, and this is thought to be due to a different spectrum of mutations in the Porphobilinogen Deaminase gene (PBGD) in the African population compared to the Caucasian population [19,20].
Sex hormones such as estrogen and progesterone have been previously implicated in the precipitation of acute AIP crises [15,21-23]. Approximately 10-30% of women with porphyria experience cyclical AIP attacks, an association thought to be secondary toprogesterone’s induction of ALA synthase, one of the major rate-limiting steps in heme synthesis [18]. This relationship of AIP with progesterone is further established by improved clinical symptoms with the use of Gonadotropin-Releasing Hormone (GnRH) agonists in patients with recurrent cyclical AIP attacks [21]. GnRH agonists decrease the number of pituitary GnRH receptors, leading to suppressed hypothalamic-pituitary-ovarian axis and decreased progesterone release [18]. Furthermore, in a Swedish population-based study, questionnaires administered to 166 women with clinical or latent AIP demonstrated that 24% of participants experienced an AIP attack with concurrent OCP use, and 18% of respondents stated that OCPs actually precipitated their first AIP attack [17]. AIP attacks can also occur during pregnancy but first presentations of AIP are rare during this time. A single case series, by Keung et al., described women with latent or undiagnosed AIP presenting with overt clinical symptoms during pregnancy or postpartum period. However, it is unclear whether the hormonal changes occurring during pregnancy alone are sufficient to exacerbate AIP, as most women described in this case series also experienced the use of a potential aggravating medication, comorbid infections, or decreased caloric intake [24].
Once the precipitating medications were removed, our patient required symptomatic treatment alone without the use of heme. Interestingly, our patient’s family did note an uncharacteristic high-carbohydrate diet. These cravings could illustrate the role of carbohydrate loading in the suppression of ALA synthase and subsequent decreased production of intermediate porphyrins. Although high carbohydrate diets have previously been advocated as integral to preventing future AIP attacks, and were commonly used in the treatment of AIP prior to heme availability, the benefit of high carbohydrate diets has not been scientifically proven [25].
Once the diagnosis of AIP is suspected and/or confirmed in a patient, it is advisable to refer the patient to one of the participating clinical centers in the Porphyrias Consortium of the Rare Disease Clinical Research Network. There are nine such clinical centers located in the USA. In addition, patients should be informed of the availability of genetic testing through the Department of Genetics & Genomic Sciences at the Icahn School of Medicine at Mount Sinai in New York, New York [26].
CONCLUSION
REFERENCES
- Anderson KE, Sassa S, Bishop DF, Desnick RJ (2001) Disorders of heme biosynthesis: X-linked sideroblasticanemias and the porphyrias. In: Scriver CR, Beaudet AL, Sly WS, et al. (eds.). The Metabolic and Molecular Basis of Inherited Disease, (8th edn), McGraw-Hill, New York, USA.
- Sack GH (1990) Acute intermittent porphyria. JAMA 264: 1290-1293.
- Solberg LA, Kahn MJ, McCrae KR (2013) Chapter 5 - Iron Metabolism, Iron Overload, and the Porphyrias. In: JeckoThachil (ed.). ASH-SAP: American Society of Hematology Self-assessment Program. (5th edn), American Society of Hematology, Washington, DC, USA.
- Bonkovsky HL, Guo JT, Hou W, Li T, Narang T et al. (2013) Porphyrin and heme metabolism and the porphyrias. Compr Physiol 3: 365-401.
- Kaushansky K, Williams WJ (2010) Chapter 57-The Porphyrias. In: Marshall Lichtman, Kaushansky k, Kipps TJ, Prchal JT, Levi MM (eds.). Williams Hematology, (8th edn), McGraw-Hill Medical, New York, USA.
- Fenske W, Allolio B (2010) The syndrome of inappropriate secretion of antidiuretic hormone: diagnostic and therapeutic advances. Horm Metab Res 42: 691-702.
- Hannon MJ, Thompson CJ (2010) The syndrome of inappropriate antidiuretic hormone: prevalence, causes and consequences. Eur J Endocrinol 162: 5-12.
- Siegesmund M, van Tuyll van Serooskerken AM, Poblete-Gutiérrez P, Frank J (2010) The acute hepatic porphyrias: current status and future challenges. Best Pract Res Clin Gastroenterol 24: 593-605.
- Kauppinen R (2005) Porphyrias. Lancet 365: 241-252.
- Pischik E, Kauppinen R (2009) Neurological manifestations of acute intermittent porphyria. Cell Mol Biol (Noisy-le-grand) 55: 72-83.
- Ventura P, Cappellini MD, Rocchi E (2009) The acute porphyrias: a diagnostic and therapeutic challenge in internal and emergency medicine. Intern Emerg Med 4: 297-308.
- 1Bloomer JR, Berk PD, Bonkowsky HL, Stein JA, Berlin NI et al. (1971) Blood volume and bilirubin production in acute intermittent porphyria. N Engl J Med 284: 17-20.
- Puy H, Gouya L, Deybach JC (2010) Porphyrias. Lancet 375: 924-937.
- Hift RJ, Thunell S, Brun A (2011) Drugs in porphyria: From observation to a modern algorithm-based system for the prediction of porphyrogenicity. Pharmacol Ther 132: 158-169.
- Stein P, Badminton M, Barth J, Rees D, Stewart MF; British and Irish Porphyria Network (2013) Best practice guidelines on clinical management of acute attacks of porphyria and their complications. Ann Clin Biochem 50: 217-223.
- American Porphyria Foundation (2014) Drug database. American Porphyria Foundation, Houston, Texas, USA.
- Bonkovsky HL, Maddukuri VC, Yazici C, Anderson KE, Bissell DM, et al. (2014) Acute porphyrias in the USA: features of 108 subjects from porphyrias consortium. Am J Med 127: 1233-1241.
- Andersson C, Innala E, Bäckström T (2003) Acute intermittent porphyria in women: Clinical expression, use and experience of exogenous sex hormones. A population-based study in northern Sweden. J Intern Med 254: 176-183.
- Robreau-Fraolini AM, Puy H, Aquaron C, Bogard C, Traore M, et al. (2000) Porphobilinogen deaminase gene in African and Afro-Caribbean ethnic groups: mutations causing acute intermittent porphyria and specific intragenic polymorphisms. Hum Genet 107: 150-159.
- Hift RJ, Meissner PN (2005) An analysis of 112 acute porphyric attacks in Cape Town, South Africa: Evidence that acute intermittent porphyria and variegate porphyria differ in susceptibility and severity. Medicine (Baltimore) 84: 48-60.
- Innala E, Bäckström T, Bixo M, Andersson C (2010) Evaluation of gonadotropin-releasing hormone agonist treatment for prevention of menstrual-related attacks in acute porphyria. Acta Obstet Gynecol Scand 89: 95-100.
- De Block CE, Leeuw IH, Gaal LF (1999) Premenstrual attacks of acute intermittent porphyria: hormonal and metabolic aspects - a case report. Eur J Endocrinol 141: 50-54.
- Dos Santos AR, De Albuquerque RR, Doriqui MJ, Costa GC, Dos Santos AP (2013) Biochemical and hematological analysis in acute intermittent porphyria (AIP): a case report. An Acad Bras Cienc 85: 1207-1214.
- Keung YK, Chuahirun T, Cobos E (2000) Acute intermittent porphyria with seizure and paralysis in the puerperium. J Am Board Fam Pract 13: 76-79.
- García-Diz L, Murcia MA, Gris JL, Pons A, Monteagudo C, et al. (2012) Assessing nutritional status of acute intermittent porphyria patients. Eur J Clin Invest 42: 943-952.
- Rare Disease Clinical Research Network. The Porphyrias Consortium.
Citation: Valente JR, Mody MD, Ramonell RP, Yin M, McClung RR et al. (2016) Severe Acute Hyponatremia as an Initial Presentation of Acute Intermittent Porphyria Triggered by a Subdermal Etonogestrel Implant. J Nephrol Renal Ther 2: 006.
Copyright: © 2016 Jean Wheeler, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.