Introduction
The balance between protein phosphorylation and dephosphorylation regulates almost every aspect of biological processes [1]. In contrast to protein phosphorylation mediated by a vast number of protein kinases, only a small number of protein phosphatases have been reported [2]. Protein phosphatases are classified into three subgroups, namely serine/threonine, tyrosine, or dual specific phosphatases [3]. The serine/threonine protein phosphatases, which include Protein Phosphatase 1 (PP1), Protein Phosphatase 2A (PP2A), and Protein Phosphatase 2B (PP2B, also called calcineurin), account for ~98% of the phosphatase activities in the mammalian heart [4]. A common feature for serine/threonine phosphatases is that they are holoenzymes composed of both catalytic subunits for the enzymatic activity and regulatory subunits that are either targeting proteins or substrates [4]. The three catalytic subunits of PP1s (PP1α, PP1β, and PP1γ), encoded by three distinct genes, are highly homologous in their catalytic domains. Their divergent N- and C-termini are hypothesized to underlie the target selectivity [5-7].
Sympathetic stimulation upon catecholamine binding generates cAMP in the heart that activates Protein Kinase A (PKA). PKA phosphorylates many Ca2+ handling and myofilament proteins, such as Ryanodine Receptor 2 (RyR2), Phospholamban (PLB), Myosin Light Chain 2 V (MLC2V), and myosin binding protein C [8-11]. Phosphorylation of SR protein PLB at serine 16 by PKA or threonine 17 by Calcium-Calmodulin-Dependent Protein Kinases (CAMKs) increases the ATPase activity of SR Ca2+ transport ATPase (SERCA) to enhance Ca2+ recycling into the SR [12]. In heart failure patients, PP1 activities were found to be increased, suggesting PP1s might be the therapeutic targets to treat heart failure patients. Consistent with this notion, Adeno-Associated Virus (AAV) 9 mediated-overexpression of phosphatase 1inhibitor-1 (I-1) improved cardiac function, while overexpression of a catalytic PP1α isoform in mice led to reduced cardiac function and heart failure [13,14]. However, cardiac specific deletion of PP1β led to heart failure and cardiac remodeling in mice [15]. These findings extended the functional complexity of PP1 isoforms in the heart, possibly due to their subcellular locations such as SR and nucleus [15-17].
Histone Deacetylases (HDACs) are a group of enzymes that remove the acetyl groups from lysine residues of a variety of target proteins including histone and non-histone substrates [18,19]. In mammalian cells, 18 HDACs have been identified and grouped into 5 different subgroups I, IIa, IIb, III, IV. In the heart, group IIa HDACs (HDAC4, HDAC5, HDAC7, and HDAC9) have been extensively studied and shown to be the endogenous inhibitors for cardiac hypertrophy [18]. For example, overexpression of HDAC4, HDAC5, and HDAC9 suppressed Myocyte Enhancer Factor 2 (MEF2) mediated gene expression to prevent pathological stimuli-induced cardiac hypertrophy [20-22]. Loss of either HDAC5 or HDAC9 led to cardiac hypertrophy in mice [21,23]. Moreover, class II HDACs are transcriptional repressors at resting state. Upon stimulation, phosphorylation of class II HDACs led to their nuclear export, resulting in the de-repression of their targeting genes [17,24-26]. However, the exact protein phosphatases that regulate the phosphorylation status of HDACs have yet to be explored.
In the present study, we hypothesized that PP1s are localized differently in the cells to regulate the phosphorylation of their substrates. In addition, we determined whether PP1s play a role in pressure-overload induced cardiac hypertrophy, and their levels during postnatal growth of the heart.
Materials and Methods
Mice husbandry and transverse aortic constriction surgery
All animal experimentation was approved by the Institutional Animal Care and Use Committee at Grand Valley State University (Protocol Number: 17-02-A). Mice were housed in standard rodent cages, and observed daily for their wellbeing such as physical activity and food/water intake. The procedures for pressure overload by Transverse Aortic Constriction (TAC) to induce cardiac hypertrophy have been previously described [15]. In brief, 8-week-old sex-matched mice were anesthetized by 1.5% isoflurane to expose the transverse aortas through a thoracotomy procedure. Constriction was performed with 7-0 silk (Ethicon, Somerville, NJ) around a 27-guage wire which was slowly removed after the constriction. After the surgery, mice were given pain medicine buprenex (0.1 mg/kg), and transferred to 30ºC chambers for overnight recovery before being transferred back to the standard housing. After 2 weeks of TAC, Doppler echocardiography was first applied to the mice to determine pressure gradients across the constrictions to ensure similar stimulation for all the mice. Cardiac function of the mice was analyzed by echocardiography using a SONOS 5500 instrument (Hewlett-Packard, Palo Alto, CA) and a 15-MHz transducer. Left ventricular Fractional Shortening percentage (FS%) was calculated using Left Ventricle Internal Diameters at the end of systole (LVIDs) and diastole (LVIDd) based on the formula: ([LVIDd-LVIDs]/LVIDd) ×100 (%). At the end of the echocardiographic analysis, mice were euthanatized by CO2 inhalation for 5 minutes followed by cervical dislocation before the hearts were removed for the weight measurement. Hearts were then fixed overnight in 10% formalin-containing phosphate buffered saline and dehydrated for embedding with paraffin. Serial 5-μm heart sections were stained with Masson’s trichrome to detect interstitial fibrosis shown as the blue staining. Fibrosis was quantified as percentage of blue staining of the tissue section using ImageJ.
Isolation of cardiomyocytes and fibroblasts from neonatal and adult hearts
Neonatal rat hearts were digested with 0.01% trypsin (Worthington, code TRLS, Lakewood, NJ) in sterile calcium and magnesium-free Hank's Balanced Salt Solution (HBSS) for 16~20 hours at 4°C with gentle rotation. The next day, trypsin inhibitors (Worthington, code SIC, Lakewood, NJ) were added to the digestion tube and incubated for 15 minutes at 37°C to stop the trypsin activity. Collagenases of 1,500 units (Worthington, code CLSPA, Lakewood, NJ) were then added to the digestion tube for an additional digestion for 60 minutes. Cells were collected through a cell strainer, and plated into 0.01% gelatin-coated dishes with 5% fetal bovine serum containing medium M199 (Corning, 10-060-CV, Corning, NY). After 60 minutes of culture, myocytes were washed off the plates and cultured into new gelatin-coated dishes, while the cells attached to the plates were considered as non-myocytes/fibroblasts.
Isolation of adult ventricular myocytes and fibroblasts has been described previously [27]. Hearts were surgically removed from 2-month-old mice after treatment with heparin (0.35 units) under anesthesia (Nembutal, 100mg/kg), and cannulated for retrograde perfusion with a solution containing liberase blendzyme (Roche, 05401151001, Indianapolis, IN). Heart tissues were dissociated by gentle pipetting and transferred to a new tube. Myocytes were allowed to settle by gravity followed by CaCl2 re-introduction. The cells in the pellet were considered as myocytes, while those in the supernatant were considered as non-myocytes/fibroblasts. After examination under the light microscope for their shape and quantity, cells were harvested immediately for RNA analysis.
RNA isolation and real-time PCR
Total RNA was purified from myocytes or fibroblasts using the RNeasy Fibrous Tissue Kit (Qiagen, 74704, Hilden, Germany) and quantified with a NANODROP 2000 Spectrophotometer (Thermo Scientific, Waltham, MA). cDNA synthesis was performed using the SuperScript III First-Strand Synthesis Kit (Invitrogen, 18080-051, Carlsbad, CA) on a regular PCR machine. Real-time PCR analysis was performed using SYBR green dye (Bio-Rad, 172–5274, Hercules, CA) on a CFX96 real-time PCR detection system (Bio-Rad, Hercules, CA). The primers to determine the mRNA level of PP1s were as follows: PP1α, forward: 5′-cctccagagagcaactacctcttc-3′, reverse: 5′-acgtcttccacagtttgatgttgt-3′; PP1β, forward: 5′-aatatggaggttttccaccagaag-3′, reverse: 5′-attgatgctagcacactcatggtt-3′; PP1γ, forward: 5′-tcttcctcagtcagcctatccttt-3′, reverse: 5′-ctccggatacttgattttgtaggc-3′; ribosomal protein 27 as the internal control, forward: 5′-ggacgctactccggacgcaaag-3’, reverse: 5′-cttcttgcccatggcagctgtcac-3’.
Nuclear fractionation and Western blot
Procedures for nuclear fractionation of cell culture has been previously described [27]. in brief, neonatal rat cardiac myocytes or HEK293 cells were collected into hypotonic buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.4% IGEPAL and protease inhibitors) and were then lysed on ice for 10 minutes. After being centrifuged for 5 minutes at 15,000 x g, the supernatant was collected as cytosolic fraction and the pellet was further re-suspended into buffer B (20 mM HEPES pH 7.9, 0.4 M NaCl, 1 mM EDTA, 10% glycerol and protease inhibitors). After 2 hours of incubation on ice, proteins eluted from the pellet were collected by centrifugation and considered as nuclear proteins. Equal amount of cytosolic and nuclear proteins were separated by 10% SDS-PAGE, transferred onto PVDF membrane, blocked with 5% non-fat milk, and incubated with the following primary antibodies: Flag (Cell signaling technology, 2368, Danvers, MA), PP1α (Santa Cruz Biotechnology, sc-6104, Dallas, TX), PP1β (Millipore, 07-1217, Burlington, MA), PP1γ (Santa Cruz Biotechnology, sc-6108, Dallas, TX), Gapdh (Fitzgerald, 10-1500, Acton, MA), Lamin A/C (Cell signaling technology, 2032, Danvers, MA). Additional primary antibodies that were also used for the Western blot: HDAC7 (Santa Cruz 7 Biotechnology, sc-74563, Dallas, TX), phospho-HDAC7 (Cell signaling technology, 3424, Danvers, MA), pSer16-phospholamban (Badrilla, A010-12AP, Leeds, UK), pThr17-phospholamban (Badrilla, A010-13AP, Leeds, UK), phospholamban (Thermo Fisher, MA3-922, Waltham, MA). Goat-anti-rabbit secondary antibody (Li-COR, P/N 925-32211, Lincoln, NE) and goat-anti-mouse secondary antibody (Li-COR, P/N 925-32211, Lincoln, NE) were utilized to visualize the results.
Immunocytochemistry
Neonatal rat cardiac myocytes grown in gelatin-coated dishes were fixed for 20 minutes with 4% paraformaldehyde followed by rinses with Phosphate-Buffered Saline (PBS) for three times. Cells were permeabilized with 0.3% triton X-100 for 15 minutes, then blocked with 3% Bovine Serum Albumin (BSA) for 20 minutes. Isoform specific PP1 antibodies were added to the cells for overnight staining of endogenous PP1 proteins, which were visualized by Alexa Fluor 488 conjugated goat-anti-rabbit secondary antibody (Thermo Fisher, A11034, Waltham, MA).
Statistics
All the results were presented as mean ± SEM from at least three independent experiments. Statistical analysis was performed using Microsoft Excel using Student’s t-test for 2 group analysis or ANOVA followed by a Bonferroni post hoc test for comparison of differences across multiple groups. P<0.05 was considered statistically significant.
Result
Distinct subcellular localizations of PP1 isoforms
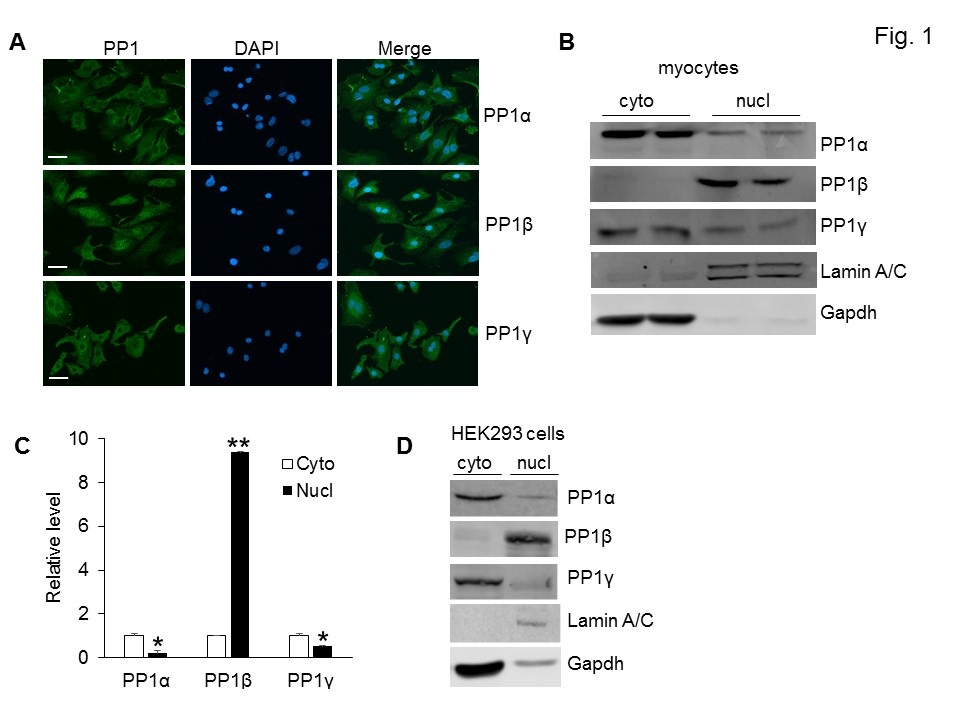
PP1 isoforms are highly homologous in the catalytic domains, but vary in the unique N and C termini, a mechanism underlying the subcellular localizations of PP1s. To further investigate the biology of PP1s in the mammalian hearts, we first compared the subcellular localizations of endogenous PP1s in neonatal cardiac myocytes using isoform specific antibodies. The majority of the PP1α and PP1γ proteins were localized in the cytoplasm, whereas PP1β was present in both the cytoplasm and the nucleus (Figure 1A). To further verify this result, we performed a nuclear fractionation experiment to compare the subcellular distribution of PP1s in neonatal cardiomyocytes. PP1β was predominantly present in the nucleus compared to PP1α and PP1γ, which also had a higher expression in the cytoplasm (Figure 1B and 1C). This result was further confirmed by biochemical fractionation of PP1 isoforms from Human Embryonic Kidney (HEK) 293 cells (Figure 1D). In both Western blots, Gapdh and Lamin A/C proteins were used as markers for cytoplasmic and nuclear proteins respectively (Figure 1B and 1D).
Distinct substrates of PP1α and PP1β in the heart
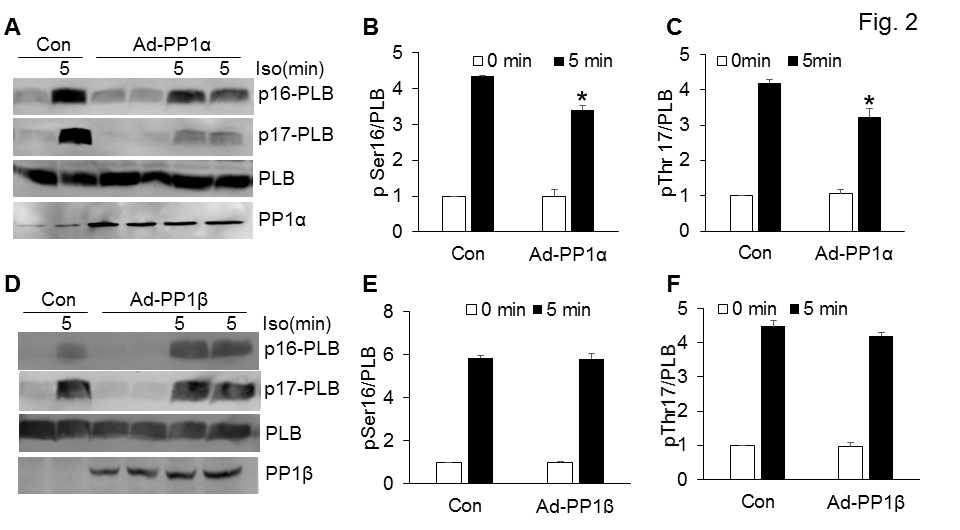
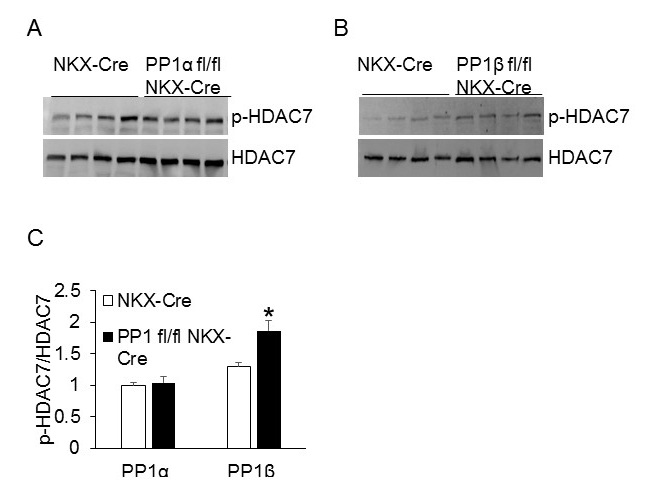
Based on the subcellular localizations of PP1 isoforms (Figure 1), we sought to investigate whether PP1s dephosphorylate different substrates in the cells. The functional role for PP1γ in the heart still remains elusive; hence PP1α and PP1β were focused for the rest of the study. We first determined whether PP1s regulate the Phosphorylation of Phospholamban (PLB), a critical protein on the SR for Ca2+ recycling. Neonatal rat cardiac myocytes were infected with adenoviruses expressing either β-gal (Con) or PP1 for 36 hours, and the PLB phosphorylation was assessed at baseline and upon isoproterenol (Iso) stimulation for 10 minutes to induce PLB phosphorylation. Overexpression of PP1α significantly reduced the phosphorylation of PLB in cells challenged with isoproterenol, an agonist for adrenergic receptors to elicit cAMP-PKA pathway (Figure 2A-2C). However, overexpression of PP1β did not alter the phosphorylation status of PLB (Figure 2D-2F). These data suggest that PP1α, the PP1 isoform localized in the cytoplasm, regulates the phosphorylation of PLB in the cytoplasm. Because PP1β was enriched in the nucleus (Figure 1B), we sought to identify the nuclear substrate for PP1β. We first assessed HDAC7, a potential target of PP1 in the nucleus [17], using PP1α (PP1α fl/flNKX-Cre) and PP1β (PP1β fl/flNKX-Cre) cardiac specific PP1 deletion mice [15]. Compared to the NKX-Cre control mice, deletion of PP1β, but not PP1α, significantly enhanced the phosphorylation of HDAC7, suggesting that PP1β is the major isoform localized in the nucleus to regulate the phosphorylation level of HDAC7 (Figure 3A-3C).
PP1 isoforms are not involved in cardiac stress responses
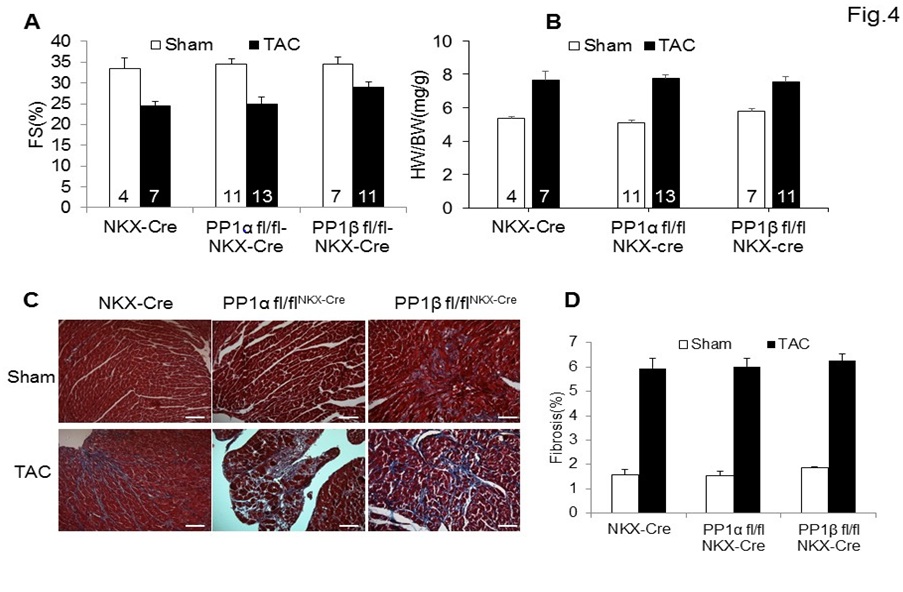
A previous study on the role of PP1 isoforms has demonstrated that cardiac specific deletion of PP1β from adult mouse hearts led to disease such as cardiac dysfunction and interstitial fibrosis [15]. However, it is still unknown whether PP1s play a role in cardiac stress responses. To address this question, we subjected 8-week-old NKX-Cre control mice or mice with cardiac specific deletion of PP1α (PP1α fl/flNKX-Cre) or PP1β (PP1β fl/flNKX-Cre) to either a sham procedure or pressure overload stimulation by Transverse Aortic Constriction (TAC). Cardiac function analysis by echocardiography demonstrated that after 2 weeks of pressure overload, there was no significant difference among all three groups of mice (Figure 4A). Similar cardiac hypertrophic growth after TAC was achieved among all three groups (Figure 4B). Moreover, histological analysis by Mason’s trichrome staining also demonstrated that deletion of individual PP1 isoforms led to similar interstitial fibrosis (blue staining) compared to the NKX-Cre control mice (Figure 4C and 4D).
PP1 levels are different in neonatal and adult hearts
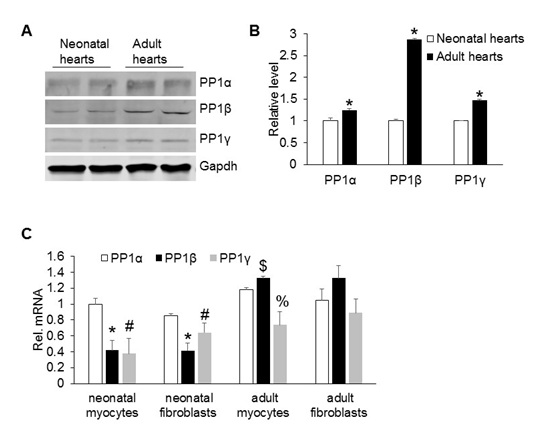
Next, we compared the expression levels of PP1s in neonatal and adult hearts to understand whether PP1s have any role in postnatal growth of the heart. The expression of all three PP1 isoforms increased in adult hearts compared to the neonatal hearts (Figure 5A and 5B). Mammalian hearts are composed of two major cell types: Myocytes, for cardiac contraction and fibroblasts that are involved in secretion of extracellular matrix proteins for tissue remodeling, such as scar formation [28]. To further determine which cell population expresses PP1s, we isolated primary cardiac myocytes and fibroblasts from neonatal and adult hearts and assessed their expression levels by real-time PCR. PP1α had a higher expression than PP1β and PP1γ in both cell types in neonatal hearts. In both cell types of adult hearts, PP1β expression was slightly higher than PP1α and PP1γ (Figure 5C).
Discussion
In this study we demonstrated that PP1 isoforms had distinct subcellular localizations to regulate the phosphorylation of specific substrates. PP1β was localized in the nucleus to regulate HDAC7 phosphorylation and PP1α dephosphorylated PLB in the cytoplasm (Figure 1-3). Our data also suggested that PP1s were not involved in the pathological cardiac hypertrophy, but might play a role in postnatal growth of the heart (Figure 4 and 5).
Numerous in vitro studies have suggested that the substrate specificity of PP1s is achieved by binding of PP1s to a large of number of structurally unrelated proteins that target them to distinct subcellular localizations [29,30]. Nearly 200 of PP1-Interacting Proteins (PIPs) have been identified in vertebrates, many of which bind to all three PP1 isoforms without isoform specificity [31]. Binding of targeting proteins to PP1s is mediated by single or combinational docking motifs on the surface of PP1s such as RVxF, RNYF, SILK [30,31]. In addition, all three PP1 isoforms demonstrate an overlapping but also distinct and dynamic localizations as well as tissue-specific expression patterns [31,32]. Further study using in vitro methods such as co-immunoprecipitation and GST-pulldown followed by mass spectrometry will help identify isoform-specific binding proteins of PP1 in the heart.
Previous studies have demonstrated that PP1and PP2A are also targeted to the mitochondria, but their specific function remains unclear [33,34]. Although we confirmed the presence of lamin A/C proteins in the nuclear fraction, we cannot rule out the possibility that proteins from other organelles such as mitochondria might be present in this crude fraction. However, we did observe a significant amount of PP1β in the nuclear fraction, deletion of which led to a moderate increase of HDAC7 phosphorylation (Figure 3), indicating that PP1β might play a role in regulating the transcriptional activity of HDAC7. Interestingly, arginine 19 in the N terminal of PP1β has been suggested for the nuclear targeting of PP1β [32].
PP1s are serine/threonine protein phosphatases that dephosphorylate a variety of substrates. Our study here showed that PP1α was localized in the cytoplasm to regulate the PLB phosphorylation, consistent with a previous study of PP1α in the heart using transgenic mouse approach [14]. PP1β seems to have functional roles in both the cytoplasm and nucleus. However, literatures regarding the role of PP1β in the cytoplasm are not consistent, possibly due to the different approaches being used. For example, using a siRNA-based approach to knock down individual PP1s from the adult cardiac myocytes, PP1β was demonstrated to be the major isoform regulating the phosphorylation of PLB [16]. Using a LoxP-Cre approach to specifically delete the PP1 isoforms from the heart, another research group demonstrated that deletion of neither PP1 isoforms influenced the phosphorylation of PLB [15]. In this study, we further demonstrated that PP1β had no effect on PLB phosphorylation (Figure 2D-2F). Instead, PP1β was localized to the nucleus to regulate HDAC7 phosphorylation. However, our study didn’t provide any mechanism on how PP1β regulates HDAC7. It is possible that dephosphorylation of HDAC7 by PP1β promotes its nuclear localization to suppress target gene expression as shown in a thymocytes study [17]. In addition, the functional role of PP1β involving gene expression through HDAC7 may also help explain the fibrotic phenotype observed in PP1β fl/flNKX-Cre mice [15].
Congenital heart disease is one of the most common development disorders that affects ~8:1000 live births worldwide [35,36]. Mutation of multiple proteins have been linked to this disease, including transcription factors NKX2-5, protein tyrosine phosphatase SHP2 [35,37]. Missense variants in PPP1Cb gene were also implicated in congenital heart disease [38]. Moreover, a recent study demonstrated greater abundance of PP1α, but unaltered levels of PP1β in heart failure patients [39]. Based on these studies, we initially hypothesized that PP1β regulates baseline cardiac function, whereas PP1α is involved in pathological responses. However, upon 2 weeks of pathological stimulation by TAC, a well-established approach in the field to induce cardiac hypertrophy, mice with loss of either PP1α or PP1β developed similar hypertrophy and dysfunction compared to the controls, suggesting PP1s are not involved in this process. It will be interesting to extend the TAC stimulation to longer period such as 14 weeks to assess whether PP1s are involved in the decompensation of the heart to failure. It is also possible that deletion of one isoform is compensated by increased level of the other two isoforms, so the overall PP1 activity is not influenced. However, even deletion of PP1β led to upregulation of both PP1α and PP1γ, mice still developed cardiac dysfunction, suggesting each PP1 isoform plays a unique function possibly due to their distinct subcellular localizations [15]. Although not involved in the cardiac stress responses, we found out that PP1s were increased during postnatal development, suggesting PP1s might play a role in postnatal growth of the heart (Figure 5A). Indeed, cardiac deletion of PP1β from fetal stage using NKX2.5-Cre led to cardiac dysfunction in mice as early as 4 weeks of age [15].
Conclusion
PP1 isoforms have distinct subcellular localizations to regulate the phosphorylation of specific substrates. PP1s are not found to be involved in cardiac stress responses, but have increased expression in postnatal growth of the heart.
Acknowledgement
This study was supported by faculty research fund at Grand Valley State University.
The authors also thank all the laboratory members for the proofreading.
Figures
Figure 1: Distinct subcellular localizations of PP1 isoforms.
(A) Immunocytochemistry analysis of endogenous PP1 isoforms in neonatal rat cardiac myocytes. Scar bars, 10 µm. (B) Western blot analysis of PP1s in the cytoplasm (cyto) and nucleus (nucl) of neonatal rat cardiac myocytes. Lamin A/C and Gapdh were used as controls for nuclear and cytosolic proteins respectively. (C) Quantification of PP1 isoforms in the cytoplasm and nucleus based on figure 1B. *P<0.05 vs cyto. ** P<0.01 vs cyto. (D) Western blot analysis of PP1s in the cytoplasm and nucleus of HEK293 cells. All the experiments were repeated 3× with similar results.
Figure 2: PP1α and PP1β selectively regulate the phosphorylation of PLB.
(A) Representative Western blot for phosphorylation of PLB at serine 16 and threonine 17, total PLB, and PP1α from neonatal rat cardiac myocytes infected with adenoviruses expressing either β-gal (Con) or PP1α. Isoproterenol (Iso) final concentration is 10 µM. (B-C) Quantification of Western blots shown in figure 2A. N=4 for each group. *P<0.05 vs Con 5 minutes. (D) Western blot for phospho-PLB at serine 16 and threonine 17, total PLB, and PP1β from neonatal rat cardiac myocytes infected with adenoviruses expressing either β-gal (Con) or PP1β. (E-F) Quantification of Western blots shown in figure 2D. N=4 for each group.
Figure 3: Cardiac-specific deletion of PP1β leads to increased HDAC7 phosphorylation.
(A-B) Western blot for phospho-HDAC7 and total HDAC7 from the adult hearts of the indicated mice at 2 months of age. This result was representative of at least two independent experiments with multiple mouse hearts. (C) Quantification of Western blots shown in figure 3A-B. N=4 for each group. *P<0.05 vs NKX-Cre.
Figure 4: Deletion of PP1α or PP1β does not influence cardiac stress responses.
(A) Echocardiographic measurements of 2-month-old mice after 2 weeks of Transverse Aortic Constriction (TAC) for Fractional Shortening (FS). Mouse numbers for each group are indicated in the bars. (B) Measurements of Heart Weight/Body Weight (HW/BW) ratios in the indicated groups of mice after 2 weeks of TAC. Mouse numbers for each group are indicated in the bars. (C) Representative histological sections stained with Masson’s trichrome (shows fibrosis in blue) from the hearts of indicated genotypes of mice. Scarbars, 50 µm.
Figure 5: Relative expression levels of PP1 isoforms in the cardiac cells.
(A) Western blot analysis for each PP1 isoform in the hearts of neonatal rats and 2-month-old mice. (B) Quantification of protein levels of PP1 isoforms in figure 5A. N=4 for each group. *P<0.05 vs neonatal hearts. (C) Real-time PCR analysis of PP1 isoform expression in cardiac myocytes and fibroblasts isolated from neonatal and adult mouse hearts. N=4 for each group. *P<0.05 vs PP1α from neonatal cells; # P<0.05 vs PP1α from neonatal cells. $ P<0.05 vs PP1α from adult myocytes; %P<0.05 vs PP1α from adult myocytes. All the data were representative of three independent experiments.