Atypical Hemolytic Uremic Syndrome Due to Neuroinfection: A Case Report and Pathophysiology Emphasis Review
*Corresponding Author(s):
Jenny L LondoñoDepartment Of Internal Medicine, Universidad El Bosque, Bogotá, Cundinamarca, Colombia
Tel:+57 3104360800,
Email:Jennitolo55@gmail.com
Abstract
TMA (Thrombotic Microangiopathy), still a challenge in the medicine practice nowadays, one of the important factor is the high suspicion needed to consider the diagnosis in a patient with multiple signs and symptoms as manifestation of one disease. In this article, based on a clinical case, we explain and offer a simple algorithm that integrates diagnosis and treatment in a in a practical and feasible manner.
A 36-year-old man, native of Venezuelan country, presented to the emergency department with complaint of malaise, bilateral ankle pain and fever for 1 week. He had medical comorbidities of mesangial proliferative glomerulonephritis and essential hypertension diagnosed 9 years ago, (being treated with nifedipine, mycophenolate and prednisone), his social history unremarkable, he denied drug abuse or smoking.
On examination he was conscious and exhibited fluctuating level of alertness. He had a blood pressure of 156/106 mmHg, pulse of 86, respiratory rate of 18, and temperature of 99°F. On precordial exam he had normal heart sounds with no murmurs, on auscultation of lungs, he had bilateral air entry with no adventitious sounds. There was no jugular venous distention. The abdominal examination result was normal. Neurological exam initially was unremarkable.
Laboratory workup showed a Complete Blood Count (CBC) with hemoglobin of 7.7g/dL, hematocrit of 22.8%, reticulocytosis of 3.2%, and a white cell count of 6.0 k/µL, platelet count was 29 k/µL. Peripheral smear was examined, which showed schistocytes (++) Drepanocytes (+) with screening test for sickle cell disease negative. Creatinine of 12.7 mg/dL (no baseline known), Blood Urea Nitrogen (BUN) 140 mg/dL, Arterial Blood Gases (ABG) with non-anion gap metabolic acidosis. A chest X-ray did not reveal any pathology, renal ultrasound findings showed changes compatible with long standing severe CKD. For the anterior data the patient was started on hemodialysis.
Several hours later patient persisted febril without any infection signs. On the second day from admission, He had a sudden episode of shaking and deviation of the head and eyes to the left, with left-beating nystagmus. During this episode, the eye movements did not cross the midline, he was responsive to questions, and appeared fully alert, despite he denied clear memories of the event.
His deteriorating mental status and inability to protect his airway required that he be placed on mechanical ventilation. Imaging studies that included a computed tomography scan of the brain showed no significant abnormalities to explain his altered mental status. He continued presenting seizure activity with decreasing interval of time, soon he was placed on complex therapy requiring benzodiazepines, valproate and the use of anesthetic therapy, after 24 hours we considered a super refractory status epilepticus condition.
In order to discard neuroinfection a lumbar puncture was indicated, and it revealed lymphocytic pleocytosis, thereby the patient was placed on antimicrobial therapy (acyclovir) considering the possibility of herpetic meningitis.
Nevertheless the most remarkable finding on this clinical case, was the simultaneous presence of schistocytes in peripheral blood, thrombocytopenia, and renal failure, leading to the suspicion of thrombotic microangiopathy. Once the ADAMTS13 level was measured (86.5%) and the absence of diarrhea history during present illness was confirmed and shiga toxin producing Escherichia coli test was negative, we concluded atypical hemolitic uremic syndrome (aHUS) as the most likely diagnosis, and the patient was as soon as possible initiated on eculizumab (after Neiserria meningitides vaccine), regardless of this effort patient died several days later (multiorgan failure), receiving just one dosis of Eculizumab.
INTRODUCTION
Thrombotic Microangiopathy (TMA), first described in 1952, is defined by the presence of fibrin and or platelet thrombi in the microcirculation of various organs [1]. Back in 2007 two clinical entities, were distinguished: Hemolytic Uremic Syndrome (HUS), characterized by predominant renal involvement, and Thrombotic Thrombocytopenic Purpura (TTP), which predominant feature was the neurological involvement [2].
In the next years, new cases appeared all over the literature, being described with overlapping symptoms, revealing the intersection of complement, coagulation, cell-mediated immunity, and state of endothelial activation, leading to a myriad of criteria and sometimes misconceptions of the pathological phenomena underlying these identities.
The present case illustrate a complex workup that is needed to accomplish, given the variable features and severity spectrum present in a broad population affected by what appeared similar clinical illness but with a totally different physiopathology understanding, that include not all ages, but both gender. Our purpose in the next lines is provided a simple understanding among the uncertainty that still holds the approach of TMA.
A SYNDROMIC APPROACH
• Vascular devices, (prosthetic heart valves, ventricular assist devices, and extracorporeal oxygenator);
• Microvascular stenosis.
In the absence of vascular devices, fragmentation of the red blood cells signifies stenosis in the arterioles and capillaries that is seen in the next 5 scenarios:
- Von Willebrand Factor (VWF) platelet thrombosis, typically observed in patients with TTP due to severe ADAMTS13 deficiency
- Platelet fibrin thrombosis, as exemplified in patients with Disseminated Intravascular Coagulopathy (DIC)
- Tumor cell invasion of the microvasculature in patients with metastatic neoplasm
- Microvascular vasculitis complicating autoimmune or certain infectious disorders
- Thrombotic microangiopathy, as observed in patients with the typical shiga toxin associated hemolytic uremic syndrome after certain Escherichia coli infection or aHUS due to defective regulation of the alternative complement pathway [3].
The major challenge comes in the distinction between TTP and aHUS, thereby the importance of the correct criteria definition, which will provide an accurate treatment and better prognosis, figure1.
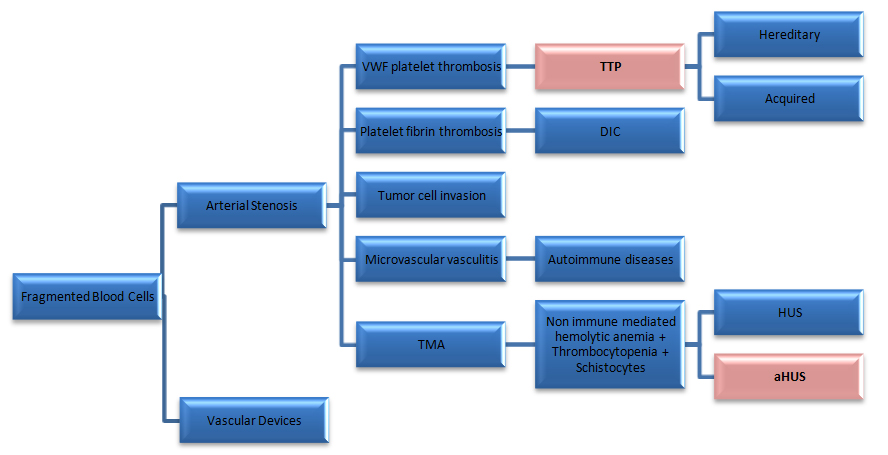 Figure 1: VWF: Von Willebrand factor; TTP: Thrombotic Thrombocytopenic Purpura; DIC: Disseminated Intravascular Coagulopathy; TMA: Thrombotic Microangiopathy; HUS: Hemolytic Uremic Syndrome; aHUS: atypical Hemolytic Uremic Syndrome.
Figure 1: VWF: Von Willebrand factor; TTP: Thrombotic Thrombocytopenic Purpura; DIC: Disseminated Intravascular Coagulopathy; TMA: Thrombotic Microangiopathy; HUS: Hemolytic Uremic Syndrome; aHUS: atypical Hemolytic Uremic Syndrome.
Classification of TMA by different mechanisms
|
VWF-platelet thrombosis due to ADAMTS13 deficiency | |
Defective regulation of complement activation | Atypical HUS |
Comorbidity as a trigger: Comorbidity as an inducer of inhibitors: |
Comorbidity as a trigger:
|
|
Other types of pathology |
|
|
|
Abbreviations: VWFV on Willebrand Factor; TTP: Thrombotic Thrombocytopenic Purpura; aHUS: Atypical Hemolytic Uremic Syndrome; HSCT: Hematopoietic Stem Cell Transplantation; CFH: Complement Factor H; MCP: Membrane Cofactor Protein; CFI: Complement Factor I; CFB: Complement Factor B; C3: Complement 3; THBD: Thrombomodulin; DIC: Disseminated Intravascular Coagulation; CAPS: Catastrophic Antiphospholipid Syndrome; HIT: Heparin Induced Thrombocytopenia; PNH: Paroxysmal Nocturnal Hemoglobinuria; VAD: Ventricular Assist Device; ECMO: Extracorporeal Membrane Oxygenation |
|
Table 1: Etiology of thrombotic microangiopathy by multiple mechanisms.
PATHOPHYSIOLOGY
aHUS - atypical Hemolytic Uremic Syndrome
Thompson and Winterborn described the relation between hypocomplementemia and HUS exposing reduced serum levels of complement C3 in patients with either sporadic or familial HUS [4]. Since then its pathophysiology is understood by an impaired regulation of complement activation, caused by reduced expression of complement regulating proteins [2].
The excessive liberation of different cleavage fragments such as C3a and C5a, and the formation of the C5b9 membrane attack complex, as a result of an impaired alternative complement pathway regulation leads to the formation of microangiopathic lesions in the kidney, C3a and C5a are anaphylatoxins that can activate platelets and endothelial cells, and mediate a proinflammatory response by their neutrophil chemotactic activity and their capacity to induce production of proinflammatory cytokines. C5b9 complexes can induce platelet activation via induction of Pselectin expression, granule secretion and a procoagulant phenotype [2].
Therefore in aHUS, microvascular stenosis may result not only from thrombosis but also directly from endothelial swelling and subendothelial expansion [3], and may be considered as a paradigm of disease resulting from inefficient protection of host endothelial cell surfaces in the setting of complement activation [2].
In up to 50% of patients with atypical HUS are found mutations in the genes that encode factor H, factor I and MCP indicating that the list of affected molecules remains incomplete, and that explains how a negative genetic or antibody test results do not exclude the diagnosis of aHUS [5], the same counts for C3 and C4 levels, that would be predicted to result in low. Although this has been reported in aHUS patients [6], additional studies suggest that no more than half of patients diagnosed with aHUS confirmed by mutation studies will demonstrate the expected low C3 and normal C4 levels, limiting their utility in the diagnosis of aHUS [5].
The following are the currently known defects in the regulation of the complement system:
- Inactivating mutations of Complement Factor H (CFH; 25%), a glycoprotein, that is synthesized in liver, macrophages, fibroblasts, endothelial cells and platelets. Factor H is the central regulator of the alternative complement pathway. It competes with factor B for binding to C3b, it accelerates the decay of C3bBb, and it acts as a cofactor for factorImediatedproteolytic inactivation of C3b
- Inactivating mutations of membrane cofactor protein (MCP or CD46: 5-10%), MCP is expressed on the surface of almost every human cell type, except erythrocytes. Together with the serine protease factor I, it degrades complement C3b and C4b that are bound to the cell surface
- Inactivating mutations of Complement Factor I (CFI), or Thrombomodulin (THBD; 5%-10%). Factor I is a soluble regulatory serine protease of the complement system that cleaves three peptide bonds in the alpha chain of C3b and two bonds in the alpha chain of C4b, thereby inactivating these proteins
- Gain of function mutations of Complement Factor B (CFB) or C3 approximately 5%-10%)
- Autoantibodies of factor H, (CFH approximately 5%-10%) [2]
Most mutations associated with atypical HUS result in defective complement control, supporting the contention that failure to inactivate or regulate C3b mediates atypical HUS development [2].
TTP - Thrombotic Thrombocytopenic Purpura
In patients with TTP, severely deficient ADAMTS13 activity has been seen in 25-79% of cases at presentation, whereas HUS is not associated with any reduction in activity or absence of ADAMTS13 [1].
Although the ADAMTS13 quantification had became a pivotal in defining diagnosis, it’s worth the effort clarify that several issues related to ADAMTS13 in TTP remain unclear. First, ADAMTS13 deficiency does not account for all cases of idiopathic TTP. Severe ADAMTS13 deficiency is detected in 13-50% of all people with TMA and in 50-90% of selected patients with TTP.This discrepancy might be due, at least in part, to different definitions of TTP being used in different studies, and to the wide range of disorders and factors associated with TTP, which include pregnancy, connective tissue diseases and drug use.
Second, neutralizing or inhibitory antibodies (which inhibit the enzymatic function of ADAMTS13 are found in 50-94% of patients in whom ADAMTS13 activity is undetectable [2]. Such nonneutralizing antibodies could enhance clearance of ADAMTS13 from the circulation or interfere with its binding to endothelial cells. The prevalence of such antibodies and their clinical relevance remain to be established.
Third, ADAMTS13 deficiency is not sufficient to induce TTP [2].
Symptoms
Gastrointestinal symptoms can commonly be seen in patients with aHUS and acquired TTP, the clinical utility of gastrointestinal symptoms to differentiate these conditions is limited. Similarly, neurologic symptoms have been used previously as a criterion to differentiate TTP from other TMAs, nonetheless severe and devastating neurologic injury has also been described in aHUS patients where the diagnosis was confirmed by the presence of complement protein mutations [7-9].
Although acute renal failure requiring kidney dialysis at presentation is a prominent clinical feature in patients with aHUS, patients with ADAMTS13-deficient TTP may also rarely present with acute renal failure. Despite renal injury in ADAMTS13- deficient TTP being common, renal failure requiring dialysis is a relatively uncommon event, occurring in 10% of patients [5].
Prognosis
The poorest prognosis is seen with CHF mutations with an up to 79% mortality or ESRD rate at 3 years and extra-renal lesions in 20% of cases, whereas in patients with MCP mutations mortality/ESRD rate is 20%. 86% of patients in the study by Caprioli and colleagues retained normal renal function and had no residual hematologic abnormalities, even after repeated disease recurrences [10,11]. Presentation, response to therapy and outcome of the disease are influenced by the genotype [11].
In 1 large retrospective study reported by Noris et al., in aHUS patients with other than MCP mutation, after 3 years of follow-up, the combined end point of ESRD or death ranged from 50% to 77%, demonstrating the poor long-term prognosis despite treatment with or without plasma exchange [6].
Treatment
Plasma exchange therapy should be started as soon as possible in all patients presenting with a suspected diagnosis of aHUS or TTP [2].
The complement system blocker eculizumab, a humanized monoclonal immunoglobulin G antibody that targets the C5 fraction of complement [13], remarkably improved response rates with a complete reversal of TMA features in more than 70% of plasma exchange-dependent or refractory aHUS, and a significant and continued improvement in renal function with dialysis discontinuation in 80% of cases.
Of note, eculizumab also provided improvement in patients with no evidence of complement abnormalities. Moreover, studies demonstrated that earlier initiation of eculizumab therapy was associated with a greater improvement in kidney outcome .
However, there is a significant risk of invasive meningococcal infection despite vaccination and antibioprophylaxis [10].
Despite the complexity of the approach of TMA, we offer a simple algorithm, that elicit key information needed in the diagnosis and treatment (Figure 2).
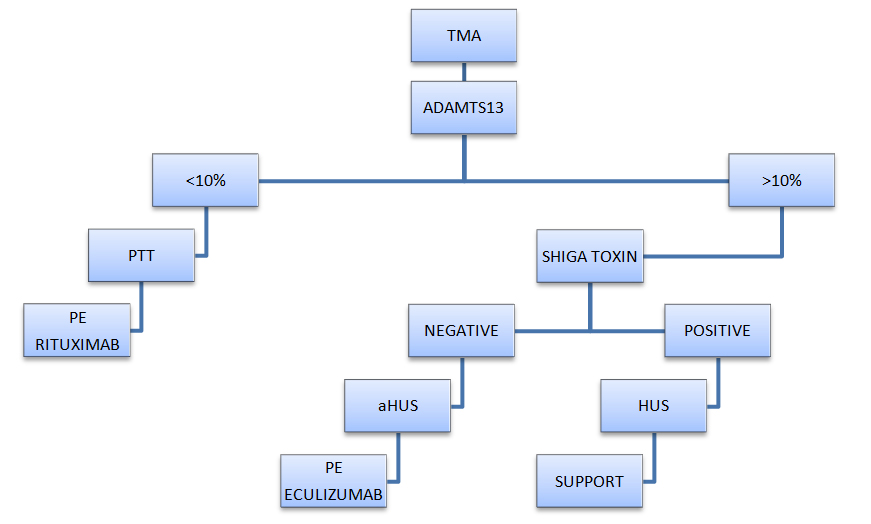
Figure 2: TMA - Diagnostic and management algorithm.
CONCLUSIONS
We presumed the neuroinfection developed aHUS as a trigger in our patient, considering a history of glomerulonephritis disease, that may be associated with complement factor H, complement factor I, and MCP mutations.
No doubt, this is an illustrative challenging clinical case, thereby a stepwise and focused review of clinical and laboratory features can greatly assist in making a definitive diagnosis in most cases.
The importance around aHUS and PTT remains in the current therapy for aHUS with eculizumab that is proved to be inversely related to the onset of the treatment.
Nevertheless the patients who fall in other identities also get benefits of a well-directed therapy, otherwise significant morbidity and mortality in both conditions overcomes.
REFERENCES
- Rafiq A, Tariq H, Abbas N, Shenoy R (2015) Atypical hemolytic-uremic syndrome: A case report and literature review. Am J Case Rep 16: 110-114.
- Fakhouri F, Frémeaux-Bacchi V (2007) Does hemolytic uremic syndrome differ from thrombotic thrombocytopenic purpura? Nat Clin Pract Nephrol 3: 679-687.
- Carey PM (2014) Thrombotic Thrombocytopenic Purpura and the Hemolytic Uremic Syndrome. Pathobiol Hum Dis A Dyn Encycl Dis Mech 27: 1595-1612.
- Thompson RA, Winterborn MH (1981) Hypocomplementaemia due to a genetic deficiency of beta1H globulin. Clin Exp Immunol 46: 110-119.
- Cataland SR, Wu HM (2014) How I treat: The clinical differentiation and initial treatment of adult patients with atypical hemolytic uremic syndrome. Blood 123: 2478-2484.
- Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, et al. (2010) Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol 5: 1844-1859.
- Noris M, Remuzzi G (2009) Atypical Hemolytic–Uremic Syndrome. N Engl J Med 361: 1676-1687.
- Gulleroglu K, Fidan K, Hançer VS, Bayrakci U, Baskin E, et al. (2013) Neurologic involvement in atypical hemolytic uremic syndrome and successful treatment with eculizumab. Pediatr Nephrol 28: 827-830.
- Salem G, Flynn JM, Cataland SR (2013) Profound neurological injury in a patient with atypical hemolytic uremic syndrome. Ann Hematol 92: 557-558.
- Tsai HM (2014) A mechanistic approach to the diagnosis and management of atypical hemolytic uremic syndrome. Transfus Med Rev 28: 187-197.
- Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, et al. (2013) Genetics of HUS?: The impact of MCP , CFH , and IF mutations on clinical presentation, response to treatment, and outcome. Blood 108: 1267-1279.
- Licht C, Ardissino G, Ariceta G, Cohen D, Cole JA, et al. (2015) The global aHUS registry: Methodology and initial patient characteristics. BMC Nephrol 16: 207.
- Jamme M, Raimbourg Q, Chauveau D, Seguin A, Presne C, et al. (2017) Predictive features of chronic kidney disease in atypical haemolytic uremic syndrome. PLoS One 12: 1-12.
Citation: Londoño JL, Zapata MI, Maiguel JA (2019) Atypical Hemolytic Uremic Syndrome Due to Neuroinfection: A Case Report and Pathophysiology Emphasis Review. Int J Case Rep Ther Stud 1: 003.
Copyright: © 2019 Jenny L Londoño, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.