Hsp90 Inhibition Affects Cell Metabolism by Disrupting Mitochondrial Protein Insertion
*Corresponding Author(s):
Maria E MycielskaDepartment Of Surgery, University Of Regensburg, Regensburg, Germany
Tel:+499419444879,
Fax:+499419444877
Email:maria.mycielska@ukr.de
Abstract
Hsp90 is a molecular chaperone interacting with hundreds client proteins. Little is known, however, about Hsp90 influence on cancer cell metabolism. Here we show that the inhibition of Hsp90 affects indirectly glycolysis by disrupting mitochondrial insertion of the elements of Translocase of the Outer Membrane (TOM) complex in tumor cells. Improper insertion of Tom40 decreases the abundance of many mitochondrial proteins resulting in reduced mitochondrial activity. This is accompanied by increased production and release of lactate and serine. These results indicate an increased rate of glycolysis with serine synthesis compensating for the loss of energy and mitochondrially derived metabolites, potentially enhancing the metastatic potential of surviving cells. Our results provide novel insights into the effects of Hsp90 inhibition on cancer cell metabolism.
Keywords
INTRODUCTION
Molecular chaperones form complexes with client proteins to assure proper folding and trafficking [1]. Heat-shock protein 90 (Hsp90) is a conserved chaperone that interacts with hundreds of clients. Cancer cells require Hsp90 to protect and keep oncoproteins in appropriate forms necessary for their activity and survival [2].
Cancer cells are known to have an altered metabolism, with glycolysis becoming their predominant means of acquiring energy even in the presence of oxygen [3]. This switch in energy production is called the "Warburg effect" [4]. In cancer cells, one of the most important enzymes in the process of glycolysis is Hexokinase II (HK II). In particular, a strong correlation between HK II expression and tumor size, differentiation, as well as tumor stage, has been shown in gastric cancer [5]. Furthermore, the malignant potential of pancreatic cancer has been correlated to the expression of HK II in the tumor tissue [6]. While HK II is responsible for metabolizing glucose into glucose-6-phosphate [7], the enzyme activity depends on its subcellular localization. HK II has greater activity when attached to the Outer Mitochondrial Membrane (OMM) through Voltage-Dependent Anion Channels (VDACs); this localization facilitates access of the enzyme to the ATP pool [8]. Interestingly, facilitated detachment of HK II from VDAC in cancer cells was found to be necessary in increasing mitochondrial activity and therefore, shifting cellular metabolism towards oxidative phosphorylation [9].
The main question of the present study was to determine the effects of blocking Hsp90 on cancer cell metabolism. Hsp90 is also present in non-transformed cells; however, its activity in non-cancerous cells was not the subject of the present study. Some reports have shown that Hsp90 does not affect glucose uptake, which would exclude a direct interaction of Hsp90 with plasma membrane glucose transporters [10]. Also, proteomic studies of the cells treated with Hsp90 inhibitors did not reveal any changes in HK II expression [11]. Hsp90 affects the assembly of elements of the mitochondrial TOM complex into mitochondrial membranes [12,13]. The latter underlies the hypothesis of the present study that disruption of the TOM complex by Hsp90 inhibition will result in a lower abundance of functional VDACs in the mitochondrial membrane and, as a consequence, in reduced glycolytic activity of the cells. Here we provide a mechanism by which blocking Hsp90 affects Tom40, VDAC and HK II abundance in mitochondria, as well as the influence of this mitochondrial imbalance on cancer cell metabolism.
MATERIALS AND METHODS
Cell culture
Transient siRNA transfection
Extraction of mitochondrial protein and determination of hexokinase activity
Western blotting and co-immunoprecipitation
For co-Immunoprecipitation (co-IP) experiments, cells were left untreated (control) or cultured in the presence of 17-DMAG (100 nM). Briefly, 500 µg of mitochondrial protein together with 1 µg of Tom40-specific antibody was mixed for 1 h at 4°C. After incubation, 25 µL of agarose beads (Santa Cruz Biotechnology) were added and samples were mixed overnight at 4°C in elution buffer from Pierce Co-Immunoprecipitation Kit (Thermo Scientific, Waltham, MA, USA). Samples were then washed 5 times with 500 µL elution buffer and protein was collected; Western blotting was performed as described previously [15,16].
Immunocytochemistry
Metabolomics
Labelled and unlabelled serine and glycine in 10 µL of media were derivatized with propyl chloroformate and analysed by HPLC-ESI-MS/MS as described previously [19]. The MRM transitions were programmed separately for unlabeled and fully labelled serine and glycine. To determine lactate release, 10 µL of medium were dried by means of a vacuum evaporator (CombiDancer, Hettich AG, Bäch, Switzerland), subjected to methoximation and silylation and analyzed by GC EI-qMS [20]. Lactate ion ratios were calculated from ions m/z 219 and 222 for unlabeled and fully labelled lactate derivative, respectively.
Animal experiments
Statistics
RESULTS
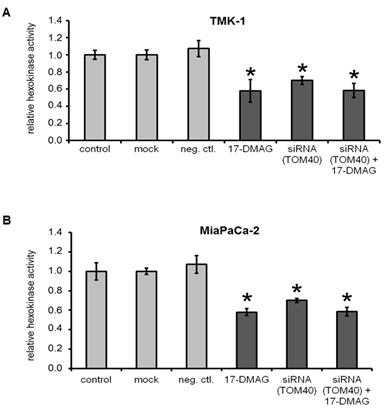
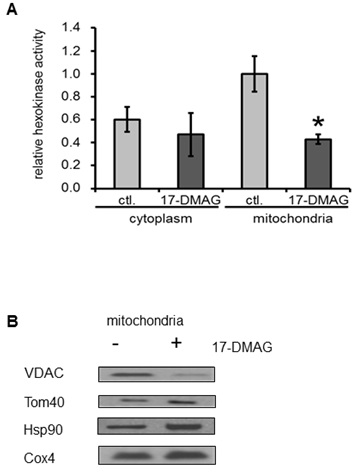
Hexokinase activity in cancer cell lines
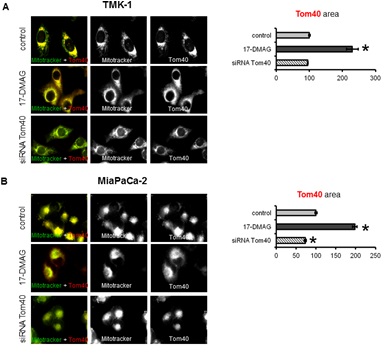
Subcellular localisation of Tom40, VDAC and HK II
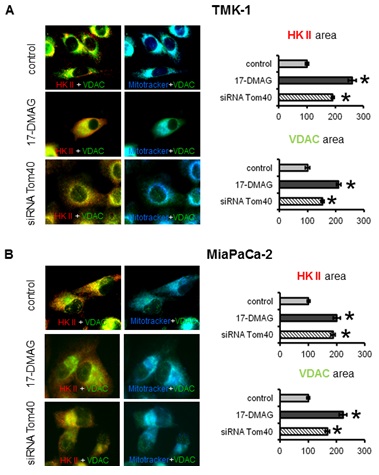
5.2.2 HK II and VDAC: TOM complex facilitates membrane transfer of mitochondrial proteins coded in the nucleus including VDACs. VDACs on the other hand bind HK II and supply ATP necessary for its activity. While our previous results describe the changes in the localization and expression of Tom40 upon Hsp90 inhibition, it was necessary to assess the consequences of these changes on subcellular localisation of VDACs and HK II. Under control conditions HK II and VDACs co-localised to mitochondria and to each other (Figure 3a, and 3b top row). Co-localization of VDACs with HK II was confirmed by the fact that the areas occupied by these two proteins were essentially the same (within a 5% difference) for TMK-1 and MiaPaCa-2 cells (Figure 3a and 3b). Upon treatment with 17-DMAG for 72 h, co-localization of VDACs with HK II was reduced (Figure 3a and 3b middle row). Accordingly, the area covered by the HK II and VDAC staining was significantly increased. This result was consistent with the changes in subcellular localisation of HK II and VDACs, and a more dispersed localisation of these proteins (less overlap with mitochondria). Interestingly, a similar effect was observed when the cells were transiently transfected with Tom40 siRNA (Figure 3a and 3b bottom row). Again, the area of staining for VDACs and HK II increased, whilst co-localization of these proteins with mitochondria was decreased. However, compared to 17-DMAG treatment, this increase was somewhat less pronounced. In conclusion, Hsp90 inhibition affects subcellular distribution of VDACs and HK II in cancer cells via a Tom40-dependent mechanism.
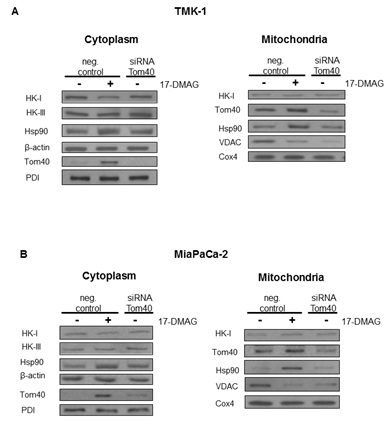
Expression level of Tom40, HKs, VDACs and Hsp90
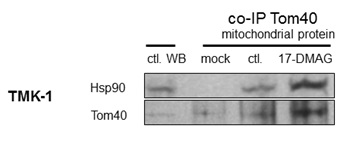
Co-immunoprecipitation of Tom40 with Hsp90
Metabolic analysis
in vivo experiments
DISCUSSION
Hsp90 plays a crucial role in oncogenesis by sustaining proper folding and trafficking of oncoproteins [2]. Therefore, Hsp90 inhibitors are studied intensively as potential anti-cancer drugs [21]. Here, we have focused on the effects of Hsp90 interactions with the mitochondrial protein Tom40. While our study does not exclude that other proteins are also involved in metabolic control through the Hsp90 inhibition, similar effects of chaperone inhibition and Tom40 knock-down do suggest Tom40 interactions with Hsp90 are a major cause of the observed metabolic alterations such as mitochondrial disruption and increase in glycolytic activity. Interestingly, the role of Hsp90 in cancer cell metabolism, in particular in glycolysis and mitochondrial respiration, has not been examined in detail. In the present study, we reveal a novel mechanism of Hsp90 action on cancer cell metabolism.
Our experiments show, that blocking Hsp90 in cancer cells results in a shift of cellular metabolism towards glycolysis, reduced Krebs cycle activity, and a change in subcellular localization of metabolism-related proteins. In the absence of changes in overall protein expression [11], we show that sub-cellular localization of HK II is substantially affected by Hsp90 blockade. This effect was the result of an altered interaction of Hsp90 with its client protein Tom40 [12,13], which is a part of the greater TOM complex [22]. Inhibition of Hsp90 resulted in higher Tom40 and Hsp90 abundance in the mitochondria. However, co-immunoprecipitation experiments showed that the observed increase was due to the presence of undissociated Hsp90-Tom40 complexes in the organelles. 17-DMAG is known to block the ATP site of Hsp90, which is needed for proper dissociation of the client protein from the chaperone [23]. Apparently, these complexes still target mitochondria but their inability to acquire proper TOM complex function triggers overexpression of Tom40 in the mitochondria and cytoplasm. This finding is consistent with the observation that ATP depletion results in Tom40 being arrested in large complexes consisting of Tom40, TOM machinery and cytosolic chaperones [12]. Therefore, our study shows, that while these Tom40-Hsp90 complexes still traffic to mitochondria, they do not dissociate in the presence of 17-DMAG. Interestingly, the use of novobiocin which is known to degrade Hsp90 client protein [24] reduces incorporation of Tom40 into the mitochondrial membrane [13]. A similar reduction was also observed when competitive Hsp90 binding was applied [12]. In all the cases, including the present research, Hsp90 disruption resulted in a reduced or faulty incorporation of Tom40 into the mitochondrial membrane. It can therefore be reasonably deduced that Hsp90 takes part in trafficking of Tom40 to the mitochondrial membrane. Interestingly, a significant role of the mitochondrial Hsp90 in keeping energy production in cancer cells has been observed recently [25,26]. In can be, therefore, assumed that blocking Hsp90 activity results in mitochondrial disruption and metabolic changes, however, the exact mechanism depends on the drug used and the part of Hsp90 affected.
Most mitochondrial proteins are coded for in the nucleus and are subsequently transferred through the mitochondrial membranes. Therefore, TOM disruption is expected to affect the abundance of several crucial molecules in this organelle, e.g. ion channels and transporters [27] that are responsible for proper exchange and communication between the mitochondria and cytoplasm. In our study we have focused on one of the major glycolytic enzymes, HK II. To acquire proper activity this enzyme needs to be attached to VDAC (believed to supply ATP for HK II; [28]), which is one of the most abundant and crucial mitochondrial communication channels [29]. We found that long-term inhibition of Hsp90 results in decreased VDAC expression in mitochondria and, in consequence, reduced abundance of HK II binding to the mitochondrial channels. We conclude, that Hsp90 inhibition affects mitochondrial import of VDACs and, thus, binding of HK II to the outer mitochondrial membrane.
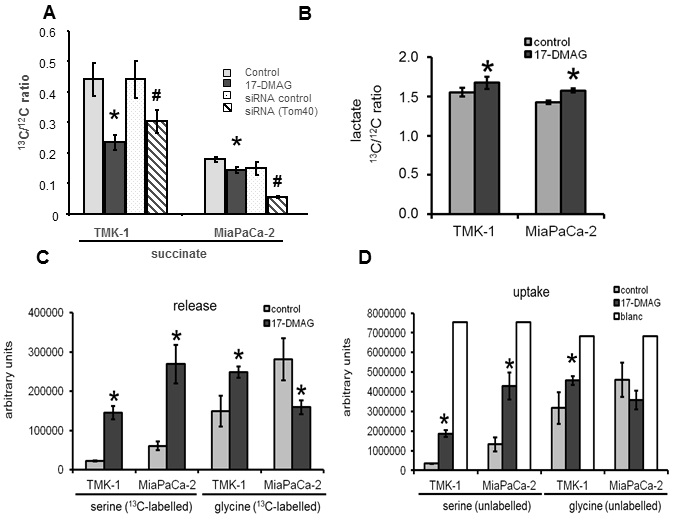
We also studied the consequences of Hsp90 inhibition-mediated changes to Tom40, HK II and VDAC on the Krebs cycle and glycolysis. Krebs cycle activity was determined as the ratio of incorporation of 13C from 13C labelled glucose into succinate, and was found to be decreased. This effect was expected because of the inability of mitochondria to properly exchange metabolites with the cytoplasm, resulting in Krebs cycle inhibition. In our research we have focused on VDACs abundance in the mitochondria in the presence of Hsp90 blocker. However, since TOM complex is involved in the import of most mitochondrial proteins, decreased mitochondrial activity is more likely related to the overall inhibited incorporation of several mitochondrial proteins including metabolic transporters such as pyruvate [30] or citrate [31]. Disruption in the abundance of mitochondrial channels and transporters could also alter mitochondrial membrane potential, additionally affecting Krebs cycle activity; this potential aspect will require further investigation.
Glycolytic activity was assessed by measuring the ratio of incorporation of 13C from 13C labelled glucose into extracellular lactate, serine and glycine. Despite the fact that HK II binding to VDACs was decreased, overall glycolytic activity was increased. There was an accumulation of lactate, serine and, in the case of the gastric cancer cell line, glycine in the cells and supernatant. The increase in lactate might be explained by the reduced utilization of pyruvate in the Krebs cycle. As a consequence, it is rather converted to lactate. Interestingly, glycolytic activity shifted mainly towards serine/glycine synthesis in both cell lines. Serine is produced by phosphoglycerate dehydrogenase from 3-phosphoglycerate, one of the intermediates of the glycolytic pathway [32]. This shift could be explained by excess serine serving as a substrate to sustain metabolic equilibrium in the cells. Importantly, serine and glycine are precursors of many biosynthetic pathways leading to the production of amino acids, purines and pyrimidines. Additionally, during the conversion of 3-phosphoglycerate to serine, glutamate is metabolized into a-ketoglutarate [32]; a-ketoglutarate, in turn, is a precursor of other metabolites (e.g. citrate) necessary for fatty acid synthesis. Moreover, serine synthesis is accompanied by NADH production needed to maintain red/ox balance. Therefore, enhancement of this serine-producing pathway via Hsp90 inhibition could partially compensate for the reduced abundance of cytosolic metabolites normally supplied by mitochondria. In fact, increased synthesis of serine and glycine derived from glycolysis has been associated with some types of cancer [33,34]. The slight differences in regulation of the serine/glycine ratio observed in the two cancer cell lines studied is likely due to the cells originating from different tissues (pancreas versus stomach).
Previous research has shown that targeting Hsp90 reduces tumor growth in vivo [10,16]; in vitro studies suggest this effect may be related to increased cell death [35]. However, we speculate from our results that although the overall cell number and tumor growth may be reduced under Hsp90 inhibition, the surviving cells will likely retain metabolic characteristics that potentially correlate with a high metastatic potential, such as increased glycolysis and serine synthesis. This finding raises concerns about Hsp90 inhibitory therapy that needs to be further researched.
CONCLUSIONS
To conclude, the present study shows that Hsp90 inhibition reduces mitochondrial respiration and increases glycolysis in the studied cancer cells, shifting metabolism mainly towards serine/glycine synthesis. These changes occur due to the disrupted interactions between Hsp90 and Tom40 indicating that Hsp90 supports mitochondrial biogenesis by assuring assembly of TOM. The overall shift towards glycolysis (a phenomenon associated with metastatic behavior of cancer cells) due to disrupted trafficking of mitochondrial proteins should be considered when applying Hsp90 inhibitors in clinics.
ACKNOWLEDGMENT
These studies were supported by the German Research Council (DFG/KFO262/La1988/3-1 and LA1988/2-1) to SAL and (DFG/KFO262/De835/2-1) to KD.
REFERENCES
- Picard D (2002) Heat-shock protein 90, a chaperone for folding and regulation. Cell Mol Life Sci 59: 1640-1648.
- Trepel J, Mollapour M, Giaccone G, Neckers L (2010) Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer 10: 537-549.
- Jin S, DiPaola RS, Mathew R, White E (2007) Metabolic catastrophe as a means to cancer cell death. J Cell Sci 120: 379-383.
- Warburg O (1956) On respiratory impairment in cancer cells. Science 124: 269-270.
- Qiu MZ, Han B, Luo HY, Zhou ZW, Wang ZQ, et al. (2011) Expressions of hypoxia-inducible factor-1a and hexokinase-II in gastric adenocarcinoma: the impact on prognosis and correlation to clinicopathologic features. Tumour Biol 32: 159-166.
- Natsuizaka M, Ozasa M, Darmanin S, Miyamoto M, Kondo S, et al. (2007) Synergistic up-regulation of Hexokinase-2, glucose transporters and angiogenic factors in pancreatic cancer cells by glucose deprivation and hypoxia. Exp Cell Res 313: 3337-3348.
- Peng Q, Zhou Q, Zhou J, Zhong D, Pan F, et al. (2008) Stable RNA interference of hexokinase II gene inhibits human colon cancer LoVo cell growth in vitro and in vivo. Cancer BiolTher 7: 1128-1135.
- Mathupala SP, Ko YH, Pedersen PL (2010) The pivotal roles of mitochondria in cancer: Warburg and beyond and encouraging prospects for effective therapies. Biochim Biophys Acta 1797: 1225-1230.
- Shulga N, Wilson-Smith R, Pastorino JG (2010) Sirtuin-3 deacetylation of cyclophilin D induces dissociation of hexokinase II from the mitochondria. J Cell Sci 123: 894-902.
- Smith-Jones PM, Solit D, Afroze F, Rosen N, Larson SM (2006) Early tumor response to Hsp90 therapy using HER2 PET: comparison with 18F-FDG PET. J Nucl Med 47: 793-796.
- Yao JQ, Liu QH, Chen X, Yang Q, Xu ZY, et al. (2010) Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin inhibits the proliferation of ARPE-19 cells. J Biomed Sci 17: 30.
- Humphries AD, Streimann IC, Stojanovski D, Johnston AJ, Yano M, et al. (2005) Dissection of the mitochondrial import and assembly pathway for human Tom40. J Biol Chem 280: 11535-11543.
- Joseph AM, Hood DA (2012) Plasticity of TOM complex assembly in skeletal muscle mitochondria in response to chronic contractile activity. Mitochondrion 12: 305-312.
- Miccoli L, Oudard S, Sureau F, Poirson F, Dutrillaux B, et al. (1996) Intracellular pH governs the subcellular distribution of hexokinase in a glioma cell line. Biochem J 313 : 957-962.
- Lang SA, Gaumann A, Koehl GE, Seidel U, Bataille F, et al. (2007) Mammalian target of rapamycin is activated in human gastric cancer and serves as a target for therapy in an experimental model. Int J Cancer 120: 1803-1810.
- Lang SA, Moser C, Gaumann A, Klein D, Glockzin G, et al. (2007) Targeting heat shock protein 90 in pancreatic cancer impairs insulin-like growth factor-I receptor signaling, disrupts an interleukin-6/signal-transducer and activator of transcription 3/hypoxia-inducible factor-1alpha autocrine loop, and reduces orthotopic tumor growth. Clin Cancer Res 13: 6459-6468.
- Mazurek MP, Prasad PD, Gopal E, Fraser SP, Bolt L, et al. (2010) Molecular origin of plasma membrane citrate transporter in human prostate epithelial cells. EMBO Rep 11: 431-437.
- Dettmer K, Nürnberger N, Kaspar H, Gruber MA, Almstetter MF, et al. (2011) Metabolite extraction from adherently growing mammalian cells for metabolomics studies: optimization of harvesting and extraction protocols. Anal Bioanal Chem 399: 1127-1139.
- van der Goot AT, Zhu W, Vázquez-Manrique RP, Seinstra RI, Dettmer K, et al. (2012) Delaying aging and the aging-associated decline in protein homeostasis by inhibition of tryptophan degradation. Proc Natl Acad Sci USA 109: 14912-14917.
- Wachsmuth CJ, Almstetter MF, Waldhier MC, Gruber MA, Nürnberger N, et al. (2011) Performance evaluation of gas chromatography-atmospheric pressure chemical ionization-time-of-flight mass spectrometry for metabolic fingerprinting and profiling. Anal Chem 83: 7514-7522.
- Mahalingam D, Swords R, Carew JS, Nawrocki ST, Bhalla K, et al. (2009) Targeting HSP90 for cancer therapy. Br J Cancer 100: 1523-1529.
- Rapaport D (2005) How does the TOM complex mediate insertion of precursor proteins into the mitochondrial outer membrane? J Cell Biol 171: 419-423.
- Onuoha SC, Mukund SR, Coulstock ET, Sengerovà B, Shaw J, et al. (2007) Mechanistic studies on Hsp90 inhibition by ansamycin derivatives. J Mol Biol 372: 287-297.
- Donnelly A, Blagg BS (2008) Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. Curr Med Chem 15: 2702-2717.
- Chae YC, Caino MC, Lisanti S, Ghosh JC, Dohi T, et al. (2012) Control of tumor bioenergetics and survival stress signaling by mitochondrial HSP90s. Cancer Cell 22: 331-344.
- Chae YC, Angelin A, Lisanti S, Kossenkov AV, Speicher KD, et al. (2013) Landscape of the mitochondrial Hsp90 metabolome in tumours. Nat Commun 4: 2139.
- Rapaport D, Neupert W (1999) Biogenesis of Tom40, core component of the TOM complex of mitochondria. J Cell Biol 146: 321-331.
- Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, et al. (2001) Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev 15: 1406-1418.
- Shoshan-Barmatz V, Ben-Hail D (2012) VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion 12: 24-34.
- Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, et al. (2012) A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 337: 96-100.
- Zara V, Ferramosca A, Papatheodorou P, Palmieri F, Rassow J (2005) Import of rat mitochondrial Citrate Carrier (CIC) at increasing salt concentrations promotes presequence binding to import receptor Tom20 and inhibits membrane translocation. J Cell Sci 118: 3985-3995.
- DeBerardinis RJ (2011) Serine metabolism: some tumors take the road less traveled. Cell Metab 14: 285-286.
- Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, et al. (2012) Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336: 1040-1044.
- Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, et al. (2011) Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet 43: 869-874.
- Karkoulis PK, Stravopodis DJ, Margaritis LH, Voutsinas GE (2010) 17-Allylamino-17-demethoxygeldanamycin induces downregulation of critical Hsp90 protein clients and results in cell cycle arrest and apoptosis of human urinary bladder cancer cells. BMC Cancer 10: 481.
Citation: Mycielska ME, Wachsmuth CJ, Dettmer K, Wagner C, Schlitt HJ, et al. (2014) Hsp90 Inhibition Affects Cell Metabolism by Disrupting Mitochondrial Protein Insertion. J Cell Biol Cell Metab 1: 002.
Copyright: © 2014 Maria E Mycielska, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.