Unverricht-Lundborg Disease: A Case Report and Literature Review
*Corresponding Author(s):
Melike BatumDepartment Of Neurology, Celal Bayar University Medicine, Manisa, Turkey
Tel:+05 0566533589,
Email:drmelikeyaman@hotmail.com
Abstract
Unverricht-Lundborg disease is a genetically inherited condition characterized by myoclonic and tonic-clonic seizures and occasionally associated with ataxia and dementia. It is consistent with type I progressive myoclonic epilepsy. Myoclonic seizures may be segmental, fragmental, or widespread, and are usually severe. They may be increased by movement, noise, stress and emotional stimuli. Slow background activity, generalized spike wave discharges, or generalized spike wave bursts of 3-5 Hz frequency are seen on Electroencephalogram (EEG). Cranial Magnetic Resonance Imaging (MRI) investigation is initially normal; and atrophy develops in time. Unverricht-Lundborg disease is a rare syndrome and its definitive diagnosis is made genetically. We aimed to remind the syndrome by evaluating the history, clinical course, adjuvant investigations and differential diagnosis of our 25 years old male case with definitive genetic diagnosis.
Keywords
Genetics; Myoclonic epilepsy; Unverricht-Lundborg disease
INTRODUCTION
Progressive Myoclonic Epilepsy (PME) is an etiologically and clinically heterogeneous neurodegenerative syndrome associated with findings such as myoclonus, epilepsy, dementia and ataxia [1]. PME Type I (Unverricht-Lundborg disease) is an autosomal recessive disease [2]. It usually begins between the ages 6 and 16. Myoclonus is observed as the first symptom in half of the patients [3]. Myoclonus that is triggered by movement or stimuli is required for diagnosis and is present in almost half of the patients [4]. Myoclonic jerks and/or tonic-clonic seizures are observed [3]. Approximately half of the patients have dysarthria and ataxia [5]. Definitive diagnosis is made by genetic investigation. In treatment, currently used antiepileptic medications improve the quality of life [3]. Some patients are relatively less affected and may survive to advanced ages. In some patients, however, there is a fulminant course, and are lost in 2 to 10 years [6-8].Diagnosis of PME is considered based on clinical features and EEG investigations in many cases. The likelihood of validating the presumed diagnosis genetically, however, is very low. In the studies conducted in our country so far, there are only 3 Unverricht-Lundborg cases whose definitive diagnosis has been made genetically.Unverricht-Lundborg disease is a rare syndrome. We aimed to remind the syndrome by evaluating our patient with genetically definitive diagnosis.
CASE REPORT
A 25 years old male patient applied with complaints of ataxia, jerks in hands and seizures. The patient, whose clinical signs started at the age of 11, had jerking stimulated especially by light and startle. His history of failure in school performance began at the age of 16 and progressively increased. He had myoclonic jerks which were asymmetrical, usually unilateral, more prominent in the distal muscle groups, leading to falls, effecting on the daily life activities, responsive to sound, light, movement and touch, and more remarkable especially at the time of awakening.
In treatment, combination of valproic acid and lamotrigine was used firstly. Because of no benefit of the treatment a combination of valproic acid and levetiracetam was used. During the use of the latter treatment (valproic acid 1500 mg/day plus levetiracetam 2000 mg/day), it was observed that generalized tonic-clonic seizures ceased and jerks evoked by light decreased. The patient was admitted to our clinic upon increasing complaints of imbalance and frequent occurrence of the jerks independent of light. Having IQ score of 96 (based on Wechsler test), the patient was learned to be successful in the years of primary and secondary schools but to have reduced success in his lessons during the years in the high-school. His medical history revealed that his mother and father were first-degree relatives. There is no any neurologic disease in his family history. Neurological exam revealed intermediate dysarthria and ataxia. Widespread generalized epileptiform discharges were observed in interictal EEG with photic stimulation (Figure 1). Investigation of cranial MRI was normal (Figure 2). In the SEP examination, somatosensory conduction was normal and wave amplitudes were high (Figure 3). JME, Myoclonus Epilepsy Ragged-Red Fiber (MERRF), aminoacidopathies, Dentatorubral Pallidoluysian Atrophy (DRPLA), and sub-types of the progressive myoclonic epilepsies were all considered in the differential diagnosis because of the clinical features. No pathological finding was found in the laboratory investigations. Diagnosis of juvenile myoclonic epilepsy was excluded because the patient was considered to have a progressive process because of regression in his ataxia and cognition. Genetic investigation was scheduled to investigate progressive myoclonic epilepsy. Homozygous mutation was found at cysteine-B location in genetic investigation using the PCR method and it was found to be consistent with PME Type I Phenotype. Based on this, definitive diagnosis of the patient was made as Unverricht-Lundborg disease. During the follow-up period, increase was seen in his complaints of myoclonic seizures and imbalance. His treatment was ordered to include levetiracetam at a dose of 3000 mg/day, valproate at a dose of 1500 mg/day and clonazepam at a dose of 2 mg/day. During the follow-ups, a decrease in the frequency of the seizures and jerks was observed; however, there was a regression in the cognition; and an increase in the ataxia.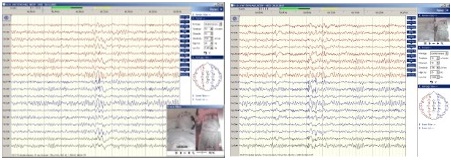
Figure 1: Widespread generalized epileptiform discharges were observed in interictal EEG with photic stimulation.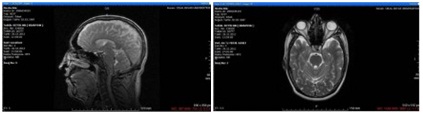
Figure 2: Kranial MRG was normal.
Figure 3: SEP examination; somatosensory conduction was normal and wave amplitudes were high.
DISCUSSION
Unverricht-Lundborg disease is an autosomal recessive neurodegenerative disease [3]. PME was first defined by Lundborg in 1903 [9]. It is common particularly in Finland and Mediterranean countries [10,11]. The PME1 gene tissue of the Unverricht-Lundborg disease is the same for the Baltic and Mediterranean types of the Unverricht-Lundborg disease, and lies in 21q22.3 region. It has been demonstrated that the Unverricht-Lundborg disease is depending on the mutation in the gene that encodes “Cystatin B” (CSTB), which is a cysteine protease inhibitor [12,13].
Common neurological signs include epileptic seizures, ataxia and cognitive dysfunction. The most common forms of seizures are myoclonic and generalized tonic-clonic seizures. Myoclonic seizures may be segmental, fragmental or widespread, and are usually severe. They may be increased by movement, sound, stress and emotional stimuli. Initial neurological exam is usually normal. Ataxia, coordination disorder, intentional tremor and dysarthria develop in 5 to 10 years from the onset of the disease. Cognitive dysfunction, slowing in mental processes over the time, emotional alterations, depression and intermediate decrease in intellectual performance are observed [14].
Our subject had dysarthria and ataxia in his neurological examination. Main symptoms included the seizures, imbalance and decreased school success. He also had jerks triggered by light and accompanied by generalized tonic-clonic seizures.
In imaging studies, the brain was found to be normal in CT and MRI investigations initially, while atrophy was observed later in the medulla, cerebellum, pons, mesencephalon, and less frequently in the cerebral hemispheres [15]. The MRI investigation was within normal limits in our subject.
Slowing in background activity, generalized spike wave discharges, and progressive generalized spike wave bursts of 3-5 Hz are observed in EEG. Rapid rhythms is observed in the frontal region [16,17].
Photosensitivity is remarkable and focal epileptiform changes are most frequently observed in the occipital region [5]. Another important point to be kept in mind about EEG is that it should be repeated at different stages of the disease because the majority of the EEG discharges may not be concurrent with the myoclonic jerks [18]. Also myoclonus in polygraphist recordings and electrographic activity association should be examined carefully. Briefly, spontaneous spike and multiple spike wave complexes, photosensitivity and slow wave activity on the background are observed in EEG investigation. Definitive diagnosis, however, is made by genetic investigation despite all of these findings [19].
Marked widespread generalized epileptiform discharges were observed in the anterior hemispheres during photic stimulation in interictal EEG investigation of our subject. On the control EEG taken one year later, the discharges were observed to be more prominent and in the form of multiple spikes.
As reported in the literature, the findings have been obtained consistent with giant SEP in our subject. In differential diagnosis, JME may be considered in the absence of action myoclonus and ataxia. The diagnosis of JME became less likely because of the progressive ataxia and the regression in his intellectual status. Other forms of PME that should be considered in the differential diagnosis include Myoclonic Epilepsy with Ragged Red Fibers (MERRF), Neuronal Ceroid Lipofuscinoses (NCL), Lafora disease and sialidosis [20]. For our case we excluded MERRF because there was no myopathy, opticatrophy, hearing loss, cardiomyopathy or peripheral neuropathy. Because of absence of visual failure we excluded NCL and sialidosis. Behavioral changes, depression, skin lesions, confusion and speech difficulties (dysarthria) are among the early signs and symptoms of Lafora disease. There was none of them in our case.
Although CSTB has been characterized in detail in vitro, its physiological function is still unknown. in vitro, CSTB binds tightly to cathepsins B, H, L and S. The main function of cathepsins is nonselective degradation of intracellular proteins to peptides and amino acids, but they also participate in antigen processing and apoptosis [21].
The most frequent mutation is the extension of the dodecamer repetition located in the area which does not make encoding of the transcription onset area of the cystatin B gene. In addition, it may also develop due to point mutations. A phenotype that is similar to the humans emerges and consists of progressive ataxia and myoclonic seizures in rats with CSTB-deficit. Pathologically, neuronal atrophy, apoptosis and gliosis are observed. The increased expressivity in the glial activation genes and apoptosis accompany this situation. Despite these developments, the physiological function and molecular pathogenesis of the CSTB gene is not known exactly [20,22-24].
Missense, nonsense, deletion and splicing mutations were also reported which affects splice sites that result in amino acid changes and predicted truncated proteins. CSTB is a cysteine protease inhibitor and controls lysosomal cathepsins [25]. Neurodegeneration has a critical role in PME1 pathogenesis and in PME1 patients’ lymphoblastoid cell lines increased catepsin activity were showed resulting in increased sensitivity to oxidative stress-induced cell death in the CSTB knock-out mice [25].
Genotype phenotype correlation has been investigated for this disease. Although individuals with pathogenic variants in CSTB gene show similar clinical phenotype and no correlation has been found between the length of the expanded dodecamer repeat and the age of onset or disease severity, recently severer phenotype was reported in compound heterozygous individuals with repeat expansion and missense mutation. In these patients age at onset of disease is lower in compound heterozygous than homozygous patients [26]. Recently it was shown that repeat numbers have a modulating effect on the age at disease onset, myoclonus severity and cortical neurophysiology [27]. In our patient we identified homozygous dodecamer repeats expansion in the promoter region of the cystatin gene by PCR-based method [28].
Main treatment of the disease is pharmacotherapy. There is no specific treatment. Valproate should be tried as the first option. Another effective drug that may be used in the early stages is levetiracetam [29]. Primidone and high-dose of piracetam are useful in myoclonic seizures. Piracetam is effective only in myoclonic seizures and it should be known that it may increase the generalized tonic-clonic seizures. Lamotrigine may be preferred especially when the side effects of valproate are to be avoided, but it occasionally increases myoclonus [30]. Clonazepam, topiramate and zonisamide may also be used in additional treatment [31].
Gabapentin, pregabalin, phenytoin, vigabatrin, carbamazepine and oxcarbazepine should be avoided because they may increase myoclonic seizures [30]. The effect of ketogenic diet is limited. Vagal and sub-thalamic deep brain stimulation has been reported to decrease the generalized seizures and myoclonus, respectively [32].
Initial treatment of our subject included a combination of valproate and lamotrigine. Upon increasing myoclonic seizures, lamotrigine was discontinued and levetiracetam was added. With the treatment of valproate at a dose of 1500 mg/day and levetiracetam at a dose of 2000 mg/day, it was observed that generalized tonic-clonic seizures disappeared and light-evoked myoclonus decreased. For myoclonic seizures, the dose of levetiracetam was increased from 2000 to 3000 mg/day, and clonazepam was added at a dose of 2 mg/day. During the follow-ups, a marked decrease was observed in the frequency of the seizures and jerks. On the neurological exam, an increase in ataxia and regression in cognition was observed.
In conclusion, although rarely observed, in case of stimulus or photosensitive myoclonus, which are observed at early ages; and in case generalized tonic-clonic seizures are accompanied by cerebellar signs and cognitive dysfunction in following years, we must consider progresive myoclonic syndromes. The diagnosis should be confirmed with genetic investigation when the disease is suspected.
REFERENCES
- [No authors listed] (1990) Classification of progressive myoclonus epilepsies and related disorders. Marseille Consensus Group. Ann Neurol 28: 113-116.
- Lehesjoki AE (2002) Clinical features and genetics of Unverricht-Lundborg disease. Adv Neurol 89: 193-197.
- Kälviäinen R, Khyuppenen J, Koskenkorva P, Eriksson K, Vanninen R, et al. (2008) Clinical picture of EPM1-Unverricht-Lundborg disease. Epilepsia 49: 549-556.
- Mazarib A, Xiong L, Neufeld MY, Birnbaum M, Korczyn AD, et al. (2001) Unverricht-Lundborg disease in a five-generation Arab family: instability of dodecamer repeats. Neurology 57: 1050-1054.
- Magaudda A, Ferlazzo E, Nguyen VH, Genton P (2006) Unverricht-Lundborg disease, a condition with self-limited progression: long-term follow-up of 20 patients. Epilepsia 47: 860-866.
- Berkovic SF, So NK, Andermann F (1991) Progressive myoclonus epilepsies: clinical and neurophysiological diagnosis. J Clin Neurophysiol 8: 261-274.
- Berkovic SF, Cochius J, Andermann E, Andermann F (1993) Progressive myoclonus epilepsies: clinical and genetic aspects. Epilepsia 3: 19-30.
- Guazzi GC, Federico A (1992) Progressive myoclonus epilepsies. Criteria for diagnosis on the basis of the follow-up of 37 cases. Acta Neurol (Napoli) 14: 469-484.
- Tassinari CA, Rubboli G, Michelucci R (1995) The epilepsy syndromes of childhood.In: Hopkins A, Shorvon S, Cascino G (eds), Epilepsy. Chapman-Hall, London. Pg no: 408-412.
- Genton P, Michelucci R, Tassinari CA, Roger J (1990) The Ramsay Hunt syndrome revisited: Mediterranean myoclonus versus mitochondrial encephalomyopathy with ragged-red fibers and Baltic myoclonus. Acta Neurol Scand 81: 8-15.
- Eldridge R, Iivanainen M, Stern R, Koerber T, Wilder BJ (1983) “Baltic” myoclonus epilepsy: hereditary disorder of childhood made worse by phenytoin. Lancet 2: 838-842.
- Lehesjoki AE, Koskiniemi M, Sistonen P, Miao J, Hästbacka J, et al. (1991) Localization of a gene for progressive myoclonus epilepsy to chromosome 21q22. Proc Natl Acad Sci USA 88: 3696-3699.
- Pennacchio LA, Lehesjoki AE, Stone NE, Willour VL, Virtaneva K, et al. (1996) Mutations in the gene encoding cystatin B in progressive myoclonus epilepsy (EPM1) Science 271: 1731-1734.
- Shields WD (2004) Diagnosis of infantile spasms, Lennox-Gastaut syndrome, and progressive myoclonic epilepsy. Epilepsia 5: 2-4.
- Moulard B, Darcel F, Mignard D, Jeanpierre M, Genton P, et al. (2003) FOunder effect in patients with Unverricht-Lundborg disease on reunion island. Epilepsia 44: 1357-1360.
- Naidu S, Niedermeyer E (1993) Degenerative disorders of the central nervous system. In: Niedermeyer E, Da Silva FHL (eds.). Electroencephalography: Basic principles, clinical applications, and related fields. Williams-Wilkins, Baltimore USA. Pg no: 360-361.
- Chippa KH, Hill RA (1997) Short-latency somatosensory evoked potentials. In: Chippa KH (ed.). Evoked Potentials in Clinical Medicine (3rdedn). Lippincott- Raven Publishers, Philadelphia. Pg no: 394-395.
- Chippa KH, Hill RA (1997) Short-latency somatosensory evoked potentials. In: Chippa KH (ed.). Evoked Potentials in Clinical Medicine (3rdedn). Lippincott- Raven Publishers, Philadelphia. Pg no: 394-395.
- Chew NK, Mir P, Edwards MJ, Cordivari C, Martino D, et al. (2008) The natural history of Unverricht-Lundborg disease: a report of eight genetically proven cases. Mov Disord 23: 107-113.
- Berkovic SF, Mazarib A, Walid S, Neufeld MY, Manelis J, et al. (2005) A new clinical and molecular form of Unverricht-Lundborg disease localized by homozygosity mapping. Brain 128: 652-658.
- Joensuu T, Lehesjoki AE, Kopra O (2008) Molecular background of EPM1-Unverricht-Lundborg disease. Epilepsia 49: 557-563.
- Polajnar M, Ceru S, Kopitar-Jerala N, Zerovnik E (2012) Human stefin B normal and patho-physiological role: molecular and cellular aspects of amyloid-type aggregation of certain EPM1 mutants. Front Mol Neurosci 5: 88.
- Lehtinen MK, Tegelberg S, Schipper H, Su H, Zukor H, et al. (2009) Cystatin B deficiency sensitizes neurons to oxidative stress in progressive myoclonus epilepsy, EPM1. J Neurosci 29: 5910-5915.
- Rinne R, Saukko P, Järvinen M, Lehesjoki AE (2002) Reduced cystatin B activity correlates with enhanced cathepsin activity in progressive myoclonus epilepsy. Ann Med 34: 380-385.
- Joensuu T, Tegelberg S, Reinmaa E, Segerstråle M, Hakala P, et al. (2014) Gene expression alterations in the cerebellum and granule neurons of Cstb(-/-) mouse are associated with early synaptic changes and inflammation. PLoS One 9: 89321.
- Koskenkorva P, Hyppönen J, Aikiä M, Mervaala E, Kiviranta T, et al. (2011) Severer phenotype in Unverricht-Lundborg disease (EPM1) patients compound heterozygous for the dodecamer repeat expansion and the c.202C>T mutation in the CSTB gene. Neurodegener Dis 8: 515-522.
- Hyppönen J, Äikiä M, Joensuu T, Julkunen P, Danner N, et al. (2015) Refining the phenotype of Unverricht-Lundborg disease (EPM1): a population-wide Finnish study. Neurology 84: 1529-1536.
- Joensuu T, Kuronen M, Alakurtti K, Tegelberg S, Hakala P, et al. (2007) Cystatin B: mutation detection, alternative splicing and expression in progressive myclonus epilepsy of Unverricht-Lundborg type (EPM1) patients. Eur J Hum Genet 15: 185-193.
- de Haan GJ, Halley DJ, Doelman JC, Geesink HH, Augustijn PB, et al. (2004) Univerricht-Lundborg disease: underdiagnosed in the Netherlands. Epilepsia 45: 1061-1063.
- Sezgin M, Baykan B (2015) Approach to the progressive myoclonic epilepsies. Guidebook of approach to the neurological diseases in the children and adolescents. Pg no: 87-93.
- Aykutlu E, Baykan B, Gürses C, Bebek N, Büyükbabani N, et al. (2005) Add-on therapy with topiramate in progressive myoclonic epilepsy. Epilepsy Behav 6: 260-263.
- Fujimoto A, Yamazoe T, Yokota T, Enoki H, Sasaki Y, et al. (2012) Clinical utility of vagus nerve stimulation for progressive myoclonic epilepsy. Seizure 21: 810-812.
Citation: Aldemir EC, Kisabay A, Batum M, Üstünoglu S, Lehesjoki AE, et al. (2016) Unverricht-Lundborg Disease: A Case Report and Literature Review. J Clin Stud Med Case Rep 3: 035.
Copyright: © 2016 Eda Cakiroglu Aldemir, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.