Inducers of Epithelial Mesenchymal Transiton and Cancer Stem Cells in Malignant Pleural Effusions
*Corresponding Author(s):
Emanuela CherubiniDepartment Of Clinical And Molecular Medicine, Sapienza University Of Rome, Rome, Italy
Tel:+39 0633775314,
Email:emanuela.cherubini@uniroma1.it
Abstract
The Epithelial to Mesenchymal Transition (EMT) plays a role not only in tumor metastasis but also in tumor recurrence. This process is believed to be tightly linked to the presence of Cancer Stem Cells (CSCs) however, it is still not clear which factors could induce EMT and how it could be a source for CSCs. It has been demonstrated that Malignant Pleural Effusion (MPEs) may represent an excellent source to identify markers and molecular mechanisms involved in EMT and CSCs development. Growth factors, cell differentiation markers and molecular adhesion are involved in some of the crucial neoplastic cell events such as proliferation, metastasis, resistance to chemotherapy and EMT.In this review, we summarize the current understanding of which molecular markers can orchestrate EMT and CSCs in MPEs.
Keywords
INTRODUCTION
Lung cancer has produced the highest mortality rate in the world, current therapy is relatively ineffective and the survival rate at 5 years is still only 15% for the advanced disease. The presence of neoplastic cells in the pleural fluid represents a common medical problem in cancer patients with advanced neoplastic disease and it leads to poor survival [1-8]. Lung and breast cancers cause approximately 75% of all MPE. However, for around 10% of MPE cases, the primary tumor is unknown [9-13]. In MPEs it has been observed that neoplastic cells produce factors that contribute to overcoming the protective mesothelial layer. For example, neoplastic cells are capable of internalizing the CD44-hyaluronan complex and hydrolyzing it in oligosaccharides showing increased permeability in the mesothelial layer and angiogenic chemotactic ability [14]. Furthermore, VEGF and bFGF produced by neoplastic cells increase the permeability of the pleural surface [15]; a low level of endostatin observed in patients with malignant pleural effusion increases endothelial cell migration, angiogenesis and tumor growth [16]. Microenvironment, hypoxia and chemokines can modify the mesothelial cell phenotype. The ability of these cells to switch dynamically between different phenotypic states led to a series of studies in which different Authors demonstrated that MPEs could be an excellent source in cancer biology investigation and the identification of potential target therapy solutions. Following this, studies identified the presence of small-sub-populations of cells, also named cancer stem cells or cancer initiating cells, within the tumor cells, causing the aggressive behaviour of cancer cells [17-19]. The presence of these sub-populations, capable of self-renewal and multipotent differentiation, could add a new element in cancer research, explain the concept of heterogeneity, relapse after treatment and resistance to conventional chemotherapies.
INDUCERS OF EMT
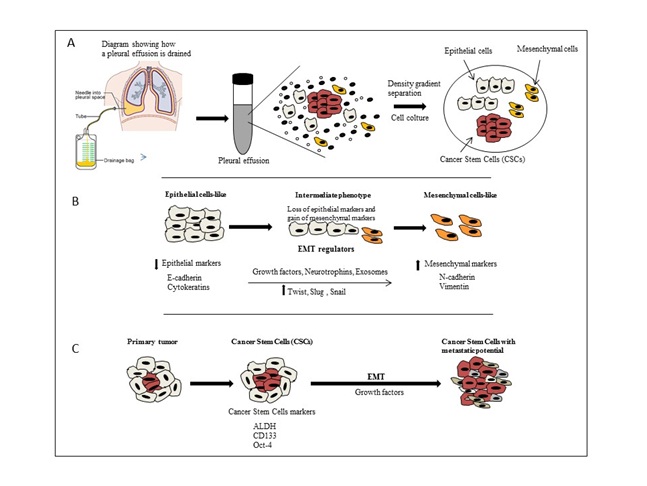 Figures A and B: The EMT mechanism transform MPE cells like from epithelial to mesenchymal cell like with suppression of epithelial and activation of mesenchymal regulator markers.
Figures A and B: The EMT mechanism transform MPE cells like from epithelial to mesenchymal cell like with suppression of epithelial and activation of mesenchymal regulator markers.Exosome
E-cadherin
Vimentin and LASP-1
Twist
Snail and slug
TGF-β
VEGF
PDGF
Neurotrophins and TrK receptors
CANCER STEM CELL MARKERS
ALDH
CD133
OCT-4
CONCLUSION
The identification of more efficient therapies for the treatment of malignant pleural effusion in patients with metastatic lung cancer is crucial to understand the mechanisms that cause current fail treatment. Characterization of CSCs in malignant pleural effusion and recent understanding of EMT contributed to better know about environment, behavior and prognosis of this tumor. Advances have been made towards elucidating causes and mechanisms of EMT in malignant pleural effusion considering EMT process as one of possible mechanisms through which CSCs are generated. However, beyond identification and characterization of cell surface markers, there is still much that remains unknown about CSCs and EMT interaction including the mechanisms they utilize to maintain their chemoresistance.
ACKNOWLEDGEMENT
This work was supported by AIRC Grant IG 17009 to R. Mancini.
REFERENCES
- Lambert MP, Herceg Z (2008) Epigenetics and cancer, 2nd IARC meeting, Lyon, France, 6 and 7 December 2007. Mol Oncol 2: 33-40.
- Uzbeck MH, Almeida FA, Sarkiss MG, Morice RC, Jimenez CA, et al. (2010) Management of malignant pleural effusions. Adv Ther 27: 334-347.
- Gompelmann D, Eberhardt R, Herth FJ (2011) Advanced malignant lung disease: what the specialist can offer. Respiration 82: 111-123.
- Kastelik JA (2013) Management of malignant pleural effusion. Lung 191: 165-175.
- Burrows CM, Mathews WC, Colt HG (2000) Predicting survival in patients with recurrent symptomatic malignant pleural effusions: an assessment of the prognostic values of physiologic, morphologic, and quality of life measures of extent of disease. Chest 117: 73-78.
- Froudarakis ME (2008) Diagnostic work-up of pleural effusions. Respiration 75: 4-13.
- Froudarakis ME (2012) Pleural diseases in the molecular era - time for more answers: introduction. Respiration 83: 2-4.
- Antony VB, Loddenkemper R, Astoul P, Boutin C, Goldstraw P, et al. (2001) Management of malignant pleural effusions. Eur Respir J 18: 402-419.
- Musani AI (2009) Treatment options for malignant pleural effusion. Curr Opin Pulm Med 15: 380-387.
- Salyer WR, Eggleston JC, Erozan YS (1975) Efficacy of pleural needle biopsy and pleural fluid cytopathology in the diagnosis of malignant neoplasm involving the pleura. Chest 67: 536-539.
- Johnston WW (1985) The malignant pleural effusion. A review of cytopathologic diagnoses of 584 specimens from 472 consecutive patients. Cancer 56: 905-909.
- Hsu C (1987) Cytologic detection of malignancy in pleural effusion: a review of 5,255 samples from 3,811 patients. Diagn Cytopathol 3: 8-12.
- Sears D, Hajdu SI (1987) The cytologic diagnosis of malignant neoplasms in pleural and peritoneal effusions. Acta Cytol 31: 85-97.
- Nasreen N, Mohammed KA, Hardwick J, Van Horn RD, Sanders K, et al. (2002) Low molecular weight hyaluronan induces malignant mesothelioma cell (MMC) proliferation and haptotaxis: role of CD44 receptor in MMC proliferation and haptotaxis. Oncol Res 13: 71-78.
- Sriram PS, Mohammed KA, Nasreen N, Hardwick J, Van Horn R, et al. (2002) Adherence of ovarian cancer cells induces pleural mesothelial cell (PMC) permeability. Oncol Res 13: 79-85.
- Nasreen N, Mohammed KA, Sanders K, Hardwick J, Van Horn RD, et al. (2003) Pleural mesothelial cell (PMC) defense mechanisms against malignancy. Oncol Res 14: 155-161.
- Basak SK, Veena MS, Oh S, Huang G, Srivatsan E, et al. (2009) The malignant pleural effusion as a model to investigate intratumoral heterogeneity in lung cancer. PLoS One 4: 5884.
- Chen SF, Lin YS, Jao SW, Chang YC, Liu CL, et al. (2013) Pulmonary Adenocarcinoma in Malignant Pleural Effusion Enriches Cancer Stem Cell Properties during Metastatic Cascade. PLoS One 8: 54659.
- Giarnieri E, De Vitis C, Noto A, Roscilli G, Salerno G, et al. (2013) EMT markers in lung adenocarcinoma pleural effusion spheroid cells. J Cell Physiol 228: 1720-1726.
- Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, et al. (2011) Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med 17: 1086-1093.
- Tang C, Ang BT, Pervaiz S (2007) Cancer stem cell: target for anti-cancer therapy. FASEB J 21: 3777-3785.
- Ma S, Chan KW, Hu L, Lee TK, Wo JY, et al. (2007) Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 132: 2542-2556.
- O'Brien CA, Pollett A, Gallinger S, Dick JE (2007) A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445: 106-110.
- Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, et al. (2005) Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 121: 823-835.
- Mancini R, Giarnieri E, De Vitis C, Malanga D, Roscilli G, et al. (2011) Spheres derived from lung adenocarcinoma pleural effusions: Molecular characterization and tumor engraftment. PLoS ONE 6: 21330
- Sodek KL, Murphy KJ, Brown TJ, Ringuette MJ (2012) Cell-cell and cell-matrix dynamics in intraperitoneal cancer metastasis. Cancer Metastasis Rev 31: 397-414.
- Allen HJ, Porter C, Gamarra M, Piver MS, Johnson EA (1987) Isolation and morphologic characterization of human ovarian carcinoma cell clusters present in effusions. Exp Cell Biol 55: 194-208.
- Sodek KL, Ringuette MJ, Brown TJ (2009) Compact spheroid formation by ovarian cancer cells is associated with contractile behavior and an invasive phenotype. Int J Cancer 124: 2060-2070.
- Casey RC, Burleson KM, Skubitz KM, Pambuccian SE, Oegema TR Jr, et al. (2001) Beta 1-integrins regulate the formation and adhesion of ovarian carcinoma multicellular spheroids. Am J Pathol 159: 2071-2080.
- Kelm JM, Timmins NE, Brown CJ, Fussenegger M, Nielsen LK (2003) Method for generation of homogeneous multicellular tumor spheroids applicable to a wide variety of cell types. Biotechnol Bioeng 83: 173-180.
- Ahmed N, Thompson EW, Quinn MA (2007) Epithelial-mesenchymal interconversions in normal ovarian surface epithelium and ovarian carcinomas: An exception to the norm. J Cell Physiol 213: 581-588.
- Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin Invest 119: 1420-1428.
- Thiery JP (2002) Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2: 442-454.
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, et al. (2008). The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133: 704-715.
- Kasper S (2009) Identification, characterization, and biological relevance of prostate cancer stem cells from clinical specimens. Urol Oncol 27: 301-303.
- Gupta GP, Massagué J (2006) Cancer metastasis: building a framework. Cell 127: 679-695.
- Sarrió D, Rodriguez-Pinilla SM, Hardisson D, Cano A, Moreno-Bueno G, et al. (2008) Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res 68: 989-997.
- Vella LJ (2014) The emerging role of exosomes in epithelial-mesenchymal-transition in cancer. Front Oncol 4: 361.
- Kim J, Kim TY, Lee MS, Mun JY, Ihm C, et al. (2016) Exosome cargo reflects TGF-β1-mediated Epithelial-to-Mesenchymal Transition (EMT) status in A549 human lung adenocarcinoma cells. Biochem Biophys Res Commun 478: 643-648.
- Bard MP, Hegmans JP, Hemmes A, Luider TM, Willemsen R, et al. (2004). Proteomic analysis of exosomes isolated from human malignant pleural effusions. Am J Respir Cell Mol Biol 31: 114-121.
- Rahman MA, Barger JF, Lovat F, Gao M, Otterson GA, et al. (2016) Lung cancer exosomes as drivers of epithelial mesenchymal transition. Oncotarget .
- Lin J, Wang Y, Zou YQ, Chen X, Huang B, et al. (2016). Differential miRNA expression in pleural effusions derived from extracellular vesicles of patients with lung cancer, pulmonary tuberculosis, or pneumonia.Tumour Biol.
- Zhao C, Li X, Su C, Li J, Cheng N, et al. (2015) High expression of E-cadherin in pleural effusion cells predicts better prognosis in lung adenocarcinoma patients. Int J Clin Exp Pathol 8: 3104-3109.
- Agiostratidou G, Hulit J, Phillips GR, Hazan RB (2007) Differential cadherin expression: Potential markers for epithelial to mesenchymal transformation during tumor progression. J Mammary Gland Biol Neoplasia 12: 127-133.
- Vasko V, Espinosa AV, Scouten W, He H, Auer H, et al. (2007) Gene expression and functional evidence of epithelial-to-mesenchymal transition in papillary thyroid carcinoma invasion. Proc Natl Acad Sci USA 104: 2803-2808.
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, et al. (2004) Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117: 927-939.
- Kang Y, Massagué J (2004) Epithelial-mesenchymal transitions: twist in development and metastasis. Cell 118: 277-279.
- Acloque H, Thiery JP, Nieto MA (2008) The physiology and pathology of the EMT. Meeting on the epithelial-mesenchymal transition. EMBO Rep 9: 322-326.
- Salvi A, Bongarzone I, Ferrari L, Abeni E, Arici B, et al. (2015) Molecular characterization of LASP-1 expression reveals vimentin as its new partner in human hepatocellular carcinoma cells. Int J Oncol 46: 1901-1912.
- D'Angelo RC, Liu XW, Najy AJ, Jung YS, Won J, et al. (2014) TIMP-1 via TWIST1 induces EMT phenotypes in human breast epithelial cells. Mol Cancer Res 12: 1324-1333.
- Yang Z, Zhang X, Gang H, Li X, Li Z, et al. (2007) Up-regulation of gastric cancer cell invasion by Twist is accompanied by N-cadherin and fibronectin expression. Biochem Biophys Res Commun 358: 925-930.
- Li J, Zhou BP (2011) Activation of β-catenin and Akt pathways by Twist are critical for the maintenance of EMT associated cancer stem cell-like characters. BMC Cancer 11: 49.
- Sasaki K, Natsugoe S, Ishigami S, Matsumoto M, Okumura H, et al. (2009) Significance of Twist expression and its association with E-cadherin in esophageal squamous cell carcinoma. J Exp Clin Cancer Res 28: 158.
- Zhang Z, Xie D, Li X, Wong YC, Xin D, et al. (2007) Significance of TWIST expression and its association with E-cadherin in bladder cancer. Hum Pathol 38: 598-606.
- Yu Q, Zhang K, Wang X, Liu X, Zhang Z (2010) Expression of transcription factors snail, slug, and twist in human bladder carcinoma. J Exp Clin Cancer Res 29: 119.
- Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, et al. (2000) The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2: 76-83.
- Hotz B, Arndt M, Dullat S, Bhargava S, Buhr HJ, et al. (2007) Epithelial to mesenchymal transition: expression of the regulators snail, slug, and twist in pancreatic cancer. Clin Cancer Res 13: 4769-4776.
- Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, et al. (2009) A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol 11: 943-950.
- Ma C, Tarnuzzer RW, Chegini N (1999) Expression of matrix metalloproteinases and tissue inhibitor of matrix metalloproteinases in mesothelial cells and their regulation by transforming growth factor-beta1. Wound Repair Regen 7: 477-485.
- Cui W, Fowlis DJ, Bryson S, Duffie E, Ireland H, et al. (1996) TGFbeta1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell 86: 531-542.
- Derynck R, Akhurst RJ, Balmain A (2001) TGF-beta signaling in tumor suppression and cancer progression. Nat Genet 29: 117-129.
- Thickett DR, Armstrong L, Millar AB (1999) Vascular endothelial growth factor (VEGF) in inflammatory and malignant pleural effusions. Thorax 54: 707-710.
- Becker PM, Alcasabas A, Yu AY, Semenza GL, Bunton TE (2000) Oxygen-independent upregulation of vascular endothelial growth factor and vascular barrier dysfunction during ventilated pulmonary ischemia in isolated ferret lungs. Am J Respir Cell Mol Biol 22: 272-279.
- Mohammed KA, Nasreen N, Hardwick J, Logie CS, Patterson CE, et al. (2001) Bacterial induction of pleural mesothelial monolayer barrier dysfunction. Am J Physiol Lung Cell Mol Physiol 281: 119-125.
- Ishimoto O, Saijo Y, Narumi K, Kimura Y, Ebina M, et al. (2002) High level of vascular endothelial growth factor in hemorrhagic pleural effusion of cancer. Oncology 63: 70-75.
- Batra H, Antony VB (2015) Pleural mesothelial cells in pleural and lung diseases. J Thorac Dis 7: 964-980.
- Mak P, Leav I, Pursell B, Bae D, Yang X, et al. (2010) ERbeta impedes prostate cancer EMT by destabilizing HIF-1alpha and inhibiting VEGF-mediated snail nuclear localization: implications for Gleason grading. Cancer Cell 17: 319-332.
- Wanami LS, Chen HY, Peiró S, García de Herreros A, Bachelder RE (2008) Vascular Endothelial Growth Factor-A stimulates Snail expression in breast tumor cells: Implications for tumor progression. Exp Cell Res 314: 2448-2453.
- Morishita Y, Ookawara S, Hirahara I, Muto S, Nagata D (2016) HIF-1α mediates Hypoxia-induced epithelial-mesenchymal transition in peritoneal mesothelial cells. Ren Fail 38: 282-289.
- Kong D, Wang Z, Sarkar SH, Li Y, Banerjee S, et al. (2008) Platelet-derived growth factor-D overexpression contributes to epithelial-mesenchymal transition of PC3 prostate cancer cells. Stem Cells 26: 1425-1435.
- Barbacid M (1994) The Trk family of neurotrophin receptors. J Neurobiol 25: 1386-1403.
- Lei L, Parada LF (2007) Transcriptional regulation of Trk family neurotrophin receptors. Cell Mol Life Sci 64: 522-532.
- Underwood CK, Coulson EJ (2008) The p75 neurotrophin receptor. The International Journal of Biochemistry & Cell Biology 40: 1664-1668.
- Li Z, Beutel G, Rhein M, Meyer J, Koenecke C, et al. (2009) High-affinity neurotrophin receptors and ligands promote leukemogenesis. Blood 113: 2028-2037.
- Nakagawara A (2001) Trk receptor tyrosine kinases: a bridge between cancer and neural development. Cancer Lett 169: 107-114.
- Ricci A, Mariotta S, Pompili E, Mancini R, Bronzetti E, et al. (2010) Neurotrophin system activation in pleural effusions. Growth Factors 28: 221-231.
- Okamura K, Harada T, Wang S, Ijichi K, Furuyama K, et al. (2012) Expression of TrkB and BDNF is associated with poor prognosis in non-small cell lung cancer. Lung Cancer 78: 100-106.
- Prakash Y, Thompson MA, Meuchel L, Pabelick CM, Mantilla CB, et al. (2010) Neurotrophins in lung health and disease. Expert Rev Respir Med 4: 395-411.
- Olgart Höglund C, de Blay F, Oster JP, Duvernelle C, Kassel O, et al. (2002) Nerve growth factor levels and localisation in human asthmatic bronchi. Eur Respir J 20: 1110-1116.
- Ricci A, Graziano P, Bronzetti E, Saltini C, Sciacchitano S, et al. (2007) Increased pulmonary neurotrophin protein expression in idiopathic interstitial pneumonias. Sarcoidosis Vasc Diffuse Lung Dis 24: 13-23.
- Dagnell C, Grunewald J, Idali F, Wikén M, Kemi C, et al. (2008) Increased levels of nerve growth factor in the airways of patients with sarcoidosis. J Intern Med 264: 463-471.
- Freund-Michel V, Frossard N (2008) The nerve growth factor and its receptors in airway inflammatory diseases. Pharmacol Ther 117: 52-76.
- Davidson B, Suo Z, Nesland JM (2004) Malignant mesothelioma. Ultrastruct Pathol 28: 179-180.
- Ricci A, De Vitis C, Noto A, Fattore L, Mariotta S, et al. (2013) TrkB is responsible for EMT transition in malignant pleural effusions derived cultures from adenocarcinoma of the lung. Cell Cycle 12: 1696-1703.
- Kupferman ME, Jiffar T, El-Naggar A, Yilmaz T, Zhou G, et al. (2010) TrkB induces EMT and has a key role in invasion of head and neck squamous cell carcinoma. Oncogene 29: 2047-2059.
- Fujikawa H, Tanaka K, Toiyama Y, Saigusa S, Inoue Y, et al. (2012) High TrkB expression levels are associated with poor prognosis and EMT induction in colorectal cancer cells. J Gastroenterol 47: 775-784.
- Jia S, Wang W, Hu Z, Shan C, Wang L, et al. (2015) BDNF mediated TrkB activation contributes to the EMT progression and the poor prognosis in human salivary adenoid cystic carcinoma. Oral Oncol 51: 64-70.
- Kim MS, Lee WS, Jeong J, Kim SJ, Jin W, et al. (2015) Induction of metastatic potential by TrkB via activation of IL6/JAK2/STAT3 and PI3K/AKT signaling in breast cancer. Oncotarget 6: 40158-40171.
- Chute JP, Muramoto GG, Whitesides J, Colvin M, Safi R, et al. (2006) Inhibition of aldehyde dehydrogenase and retinoid signaling induces the expansion of human hematopoietic stem cells. Proc Natl Acad Sci USA 103: 11707-11712.
- Moreb JS (2008) Aldehyde dehydrogenase as a marker for stem cells. Curr Stem Cell Res Ther 3: 237-246.
- Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, et al. (2007) ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1: 555-567.
- Matsui W, Huff CA, Wang Q, Malehorn MT, Barber J, et al. (2004) Characterization of clonogenic multiple myeloma cells. Blood 103: 2332-2336.
- Hiratsuka S, Watanabe A, Aburatani H, Maru Y (2006) Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol 8: 1369-1375.
- O'Brien CA, Pollett A, Gallinger S, Dick JE (2007) A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 445: 106-110.
- Yin S, Li J, Hu C, Chen X, Yao M, et al. (2007) CD133 positive hepatocellular carcinoma cells possess high capacity for tumorigenicity. Int J Cancer 120: 1444-1450.
- Horst D, Scheel SK, Liebmann S, Neumann J, Maatz S, et al. (2009) The cancer stem cell marker CD133 has high prognostic impact but unknown functional relevance for the metastasis of human colon cancer. J Pathol 219: 427-434.
- Ishigami S, Ueno S, Arigami T, Uchikado Y, Setoyama T, et al. (2010) Prognostic impact of CD133 expression in gastric carcinoma. Anticancer Res 30: 2453-2457.
- Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, et al. (2008) Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ 15: 504-514.
- Chambers I, Colby D, Robertson M, Nichols J, Lee S, et al. (2003) Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell 113: 643-655.
- Hochedlinger K, Yamada Y, Beard C, Jaenisch R. (2005) Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell 121: 465-477.
- Freberg CT, Dahl JA, Timoskainen S, Collas P (2007) Epigenetic reprogramming of OCT4 and NANOG regulatory regions by embryonal carcinoma cell extract. Mol Biol Cell 18: 1543-1553.
- Gupta PB, Chaffer CL, Weinberg RA (2009) Cancer stem cells: mirage or reality? Nat Med 15: 1010-1012.
Citation: Cherubini E, Giarnieri E, Ricci A, Mariotta S, Giovagnoli MR et al. (2016) Inducers of Epithelial Mesenchymal Transiton and Cancer Stem Cells in Malignant Pleural Effusions. J Cytol Tissue Biol 3: 011.
Copyright: © 2016 Emanuela Cherubini, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.