Centella Asiatica Extracts Regulates Aluminium Chloride-Induced Neurotoxicity in Rats: Impact on Inflammation, Apoptosis and Biogenic Amine Levels
*Corresponding Author(s):
Shaik AmjadDepartment Of Pharmacology And Environmental Toxicology, University Of Madras, Tamilnadu, India
Tel:+91 44 2539 9422,
Email:shaikamjadphd@gmail.com
Abstract
Objectives and design: Aluminium (Al) is an environmental neurotoxin that affects cerebral functions and causes health complications. The antioxidant and memory enhancing effects of Centella Asiatica (CA) are well known in the past few decades. Therefore, the present study has been designed to investigate the therapeutic potential of CA against chronic Aluminium Chloride (AlCl3) exposure induced rats. Wistar albino rats were segregated into four groups: group 1-control rats, group 2-rats received AlCl3 (300 mg/kg body weight, every day orally) for 60 days, rats in group 3-received CA (500 mg/kg body weight, orally) and group 4 rats were initiated with both AlCl3 and CA treatment.
Material and methods: Neurotoxicity was assessed by measuring brain biogenic amines, Al concentration in blood and brain, the levels of pro-inflammatory cytokines and later the apoptotic related components. Significant depletion of neither brain Dopamine (DA), Epinephrine (E), Nor Epinephrine (NE) nor 5-Hydroxytryptamine (5-HT) levels was observed following AlCl3 exposure. Administration of AlCl3 raised the pro-inflammatory cytokines levels (tumor necrosis factor alpha, TNF-α and inducible nitric oxide synthase, iNOS) and modulated the levels of apoptotic proteins cytochrome c, caspases 3, with altered Bax/Bcl-2 ratio in favour of apoptosis in cortex that are determined by western blotting and immunohistochemistry.
Results: Administration of CA significantly increased biogenic amine levels, decreased Al concentration compared to Al-induced animals. CA ameliorates AlCl3-induced inflammatory niche. Further, treatment with CA ameliorated neuronal apoptosis by reducing cytochrome c, caspase- 3 expressions significantly and by modulating Bax/Bcl-2 ratio.
Conclusion: The findings of this study suggest that CA protects the cerebral cortex against AlCl3-induced neurotoxicity. This neuroprotective effect is partially mediated by its anti-oxidant, anti-inflammatory and anti-apoptotic activities as well as elevating biogenic amine levels.
Keywords
Aluminium chloride; Apoptosis; Centella asiatica; iNOS; TNF-?
INTRODUCTION
Aluminum (Al) is a potent environmental neurotoxin and has been associated with several disorders. Al and its salts are widely exposed to human body due to their presence in several products including cosmetics, drugs, food additives (salt, spices, yellow cheese and herbs) and a powerful flocculants in water purification. Al has the potential to be toxic for humans and its compounds have neurotoxic manifestations which further shown to contribute several age-related neurodegenerative diseases [1]. Chronic exposure to Al causes pathogenesis of age-related illnesses such as the Alzheimer’s Disease (AD), the Parkinson’s disease, the Huntington’s disease, inflammation, Amyloid Beta (Aβ) deposition and plaque formation in the brain and suggested that this excess might be a possible contributing factor for the aging process by inflicting oxidative damage [2]. Evidence suggests that inhaled Al not only causes neurologic signs, which mimic progressive neurodegeneration, but also results in neurofilamentous changes in the cerebral cortex focal region where more Al accumulation takes place [3,4]. Previous studies have shown that, the excessive concentration of Al in neurotic deposits, plaques and neurofibrillary tangles in the brain of Alzheimer’s patients [5].
Al is a well-documentedan experimental neurotoxicant that enhances neuroinflammatory events in the brain by multiple mechanisms. Accumulating evidence suggests that Aluminium Chloride (AlCl3) promote free radical generation by potentiating the pro-oxidant properties of transition metals (iron and copper) [6]. In addition, AlCl3 exerts its own pro-oxidant effect by altering calcium flux and homeostasis and to facilitate peroxidation of membrane lipids and antioxidant enzymes [7]. Beside this, various Al salt complexes can alter neuronal signal transduction pathways associated with glial cell activation, which further promote an uncontrolled inflammatory cascade in the brain. These activated glial cells secrete cytotoxic agents including Reactive Oxygen Species (ROS), Nitric Oxide (NO), inflammatory cytokines Like Tumor Necrosis Factor alpha (TNF-α), inducible Nitric Oxide Synthase (iNOS) which are toxic and inturn cause synaptic abnormalities ultimately related to memory dysfunction. Earlier study has evidenced that AlCl3 activates astrocytes and glial cells which releases different inflammatory mediators due to induction of apoptosis [8,9]. Therefore, overall effects are to reduce pro-inflammatory mediator’s production by activated microglia and astrocytes should be useful for prevention of neuroinflammation and eventually neuroprotection.
Apoptosis under mitochondrial control has been implicated in a progressive and selective loss of neurons, which involve the release of cytochrome c into the cytoplasm and initiation of the apoptosis cascade. AlCl3 is a potent cholinotoxin and causes aberrant apoptotic neuronal loss by stimulating ROS production, which is a characteristic symptom of neurodegeneration [10-12]. It induces apoptosis of neurons and astrocytes via involvement of caspases activation in rodent and in vitro models [13,14]. Reactivation of caspases has been implicated in mediating neural death during pathological processes causing neuronal loss in acute and chronic neurodegenerative diseases. Caspase-induced apoptosis has been documented in Amyloid-β-induced Alzheimer's and 6-hydroxydopamine-induced Parkinson's neuronal apoptosis [15,16]. Restriction of apoptosis and its associated factors during neurodegeneration is presumed to be beneficial for the survival of neurons. Thus clinical trials are in progress that anti caspase approach in neurodegeneration will be an exciting avenue for therapeutic neuroprotective interventions [17].
The antioxidant capabilities of Centella Asiatica (CA) have been documented previously in different experimental models. Centella Asiatica (CA), also known as gotu kola and Indian pennywort, is a perennial herbaceous creeper of the Apiaceae family. Recently, CA has been reported as a neuroprotectant against different degenerative disorders. CA modulate the immune system [18], act as antioxidant [19], prevent alleviation of oxidative stress [20], act as anti-inflammatory agent [21], and inhibit proliferation of cancer cells [22]. Yet, the most widely reported health benefit of this herb is in improving the brain function, particularly related to learning and memory. In fact, CA has been reported to stimulate nerve regeneration in vitro [23], which could explain its brain protective effect. In humans, it has been reported to improve mental ability of the mentally retarded children and decrease the amyloid-β levels in the brain [24,25]. Previously, we have demonstrated that CA inhibited cognitive impairment and improves learning and memory in Al-induced rats by reducing oxidative stress and Acetyl Cholinesterase (Ach E) activity [26]. Based on this background, the present study was carried out to investigate the protective effect of CA against AlCl3 induced inflammation and oxidative stress associated mitochondria apoptosis. Therefore in this study, we have investigated the efficacy of CA in terms of biogenic amines, expression of pro-inflammatory cytokines TNF-α, iNOS and apoptotic inducing factors in cortex of rat brain.
MATERIALS AND METHODS
Animals
Preparation of the plant extract: Fresh leaves of CA were collected from regular vendor and species identification was done from Center for Advanced Studies in Botany, University of Madras, Chennai, India. The leaves were cleaned, shade dried and coarsely ground with grinder. The coarse powder of plant was extracted with 8 parts of distilled water under boiling for 5 hrs and was filtered through a 400 mesh cloth to collect the extract. The extract was concentrated and freeze dried to get a powder of greenish brown in color. The percentage w/w yield of the extract was 41%. Standardized aqueous extract of CA and AlCl3 solutions (made freshly at the beginning of each experiment daily). For oral administration, AlCl3 and CA were administrated in a dose of 0.5 ml/100 g body weight. The animals were divided into four groups (n=6). Group 1 control rats. Group 2 rats administered with AlCl3 (300 mg/kg) for 60 days. Group 3 rats received CA alone (dissolved in water, 500 mg/kg). Group 4 rats co-treated with CA and induced with AlCl3 simultaneously for 60 days. The doses of AlCl3 [26,27] and CA [26] were selected based on published report of our own laboratory.
RESULTS
Effect of CA on Al concentration in serum and brain tissue
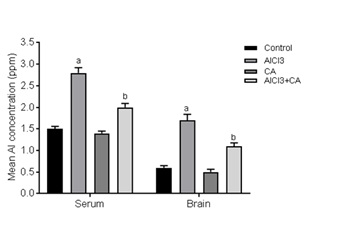
Figure 1: Effect of CA supplementation on Al concentration in serum and brain tissue of control and experimental rats subjected to AlCl3. Results were expressed as mean ± SD (n=6). Values are statistically significant at p<0.05; compared with aAlCl3 vs. control; bAlCl3+ CA vs. AlCl3.
Effect of CA on AlCl3-induced rat brain neurotransmitters
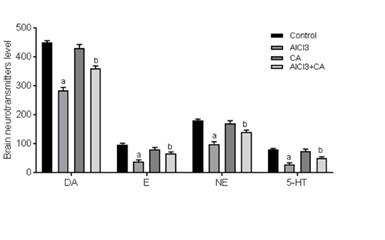
Figure 2: Graph represents the effect of CA supplementation on the levels of brain monoamine neurotransmitters in the rats of control and experimental groups. Dopamine (DA, ng/g), epinephrine (E, ng/100g), norepinephrine (NE, ng/100g) and 5-hydroxytryptamine (5-HT, ng/g). Results were expressed as mean ± SD (n=6). Values are statistically significant at p<0.05; compared with aAlCl3 vs. control; bAlCl3+ CA vs. AlCl3.
CA treatment decreases the AlCl3-induced expressions of TNF-α and iNOS
An abundant activation of iNOS (Figure 3F) positive cells was evident in cerebral cortex tissue from rats exposed to AlCl3 (p<0.05), in contrast to an almost negligible expressions was found in control and CA-alone treated animals (Figure 3E). CA-treated group of rats exhibited an evident reduction of expression of iNOS (p<0.05) (Figure 3H) when compared with AlCl3-induced and control group. No adverse changes were noticed in cortex of control and CA-alone-treated groups of rats (Figure 3G). The total number of immunoreactive cells was quantified and is represented in graph (Figure 3I).
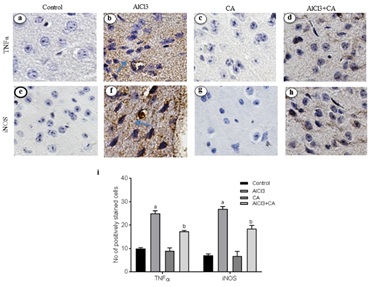
Figure 3: Representative micrographs of immune stained specimens of cortex from control and experimental groups of rats. Brain specimens were probed with antibody specific for TNFα and iNOS. a) TNF-α expression in cortex of control group; b) TNF-α expression in cortex of AlCl3-induced group; c) CA alone (drug control) group; d) TNF-α expression in cortex of AlCl3-induced and CA treated group; e) iNOS expression in cortex of control group; f) iNOS expression in cortex of AlCl3-induced group; g)CA alone (drug control) group; h) iNOS expression in cortex of AlCl3-induced and CA treated group; Scale bar 100 ?m, magnification ×20. (i) Immunoreactivity was assessed and measured objectively by two independent observers. For quantitation, data expressing the respective protein expression were quantitated in ten fields/section and an average was used to denote the no. of positively stained cells and is represented in graph. Arrows indicate the positively stained cells. Hypothesis testing method included two-way Analysis of Variance (ANOVA) followed by Least Significant Difference (LSD). Results are given as statistically significant at p<0.05; compared with aAlCl3 vs. control; bAlCl3+ CA vs. AlCl3.
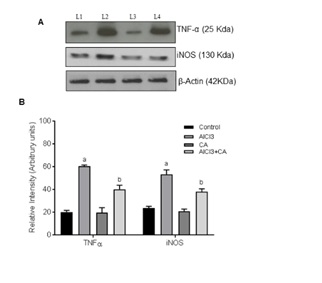
Figure 4: Representative immunoblots of TNF-α and iNOS protein expressions in the cortex of control and experimental groups of rats. Western blot analyses were performed using probes specific for TNF-α, iNOS, and β-actin as indicated. Lane 1 Control; Lane 2 AlCl3-induced; Lane 3 CA alone; Lane 4 AlCl3-induced and CA treated. Data expressing the corresponding protein levels were assessed using densitometry and is expressed in relative intensity (arbitrary units). Hypothesis testing method included two-way Analysis of Variance (ANOVA) followed by Least Significant Difference (LSD). Results are given as statistically significant at p<0.05; compared with aAlCl3 vs. control; bAlCl3+ CA vs. AlCl3.
CA treatment suppressed cytochrome c expression in AlCl3-induced rat brain
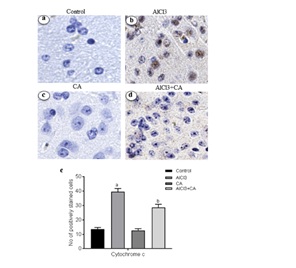
Figure 5: (A) Representative micrographs of immune stained sections of cytochrome c expression in cortex from control and experimental groups of rats. Brain specimens were probed with antibodies specific for cytochrome c (Magnification ×40). Immunoreactivity was assessed and measured objectively by two independent observers. B) For quantitation, data expressing total number of positively stained cells quantification were performed by counting the positively stained cells in 10 fields/section by two independent observers in blinded fashion, and the average was used to denote the total number of positively stained cells. Hypothesis testing method included two-way Analysis of Variance (ANOVA) followed by Least Significant Difference (LSD). Results are given as statistically significant at p<0.05; compared with aAlCl3 vs. control; bAlCl3+ CA vs. AlCl3.
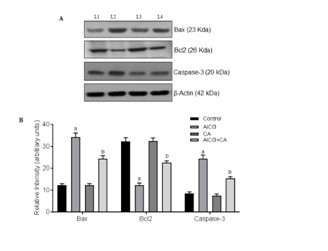
CA treatment suppressed apoptotic-inducing proteins in AlCl3-induced rat brain
DISCUSSION
Al is a ubiquitous metal and implicated in the pathophysiology of neurodegenerative diseases. Reports suggest that AlCl3 alters cytoskeletal dynamics of neurons and astrocytes leading to pathogenesis of neurodegeneration. In mammals, Al accumulation in the brain has been linked to neurodegenerative diseases such as PD and AD [4, 35]. Understanding the etiology of neurodegeneration and identifying ways to achieve early prevention are being considered as important approaches in the management of neurological diseases. Counteracting attempts to prevent/inhibit neurodegeneration and enhance neuronal survival has been greatly summoned. Recently, the traditional medicinal plant CA and its extracts have gained much interest for their ability to modulate neuronal function and influence learning and memory processes [36,37]. Hence, the present study was an attempt to understand the reduction of neuroinflammatory signals and apoptotic mechanisms underlying neuroprotective efficacy of CA in a rat model of neurodegeneration.
Brain is considered a preferential site of Al accumulation mainly at some cortical regions and hippocampus via crossing the Blood-Brain Barrier (BBB) [38]. The obtained data demonstrated that the serum Al level was elevated in Al exposed rats, indicated an occurrence of serum hyper-aluminemia which resulted in an elevation of brain tissue Al content. This elevation appears to be a consequence of an alteration in the permeability of the BBB, it has been shown that Al somehow interacts with the BBB via interfering with its function at the endothelial cell level, whereas positively charged Al ions neutralize the anionic sites of the endothelial cell surface and also may consequently altering the function of the BBB [39]. Concomitant treatment with CA was associated with a significant reduction in serum and brain Al content this depletion may be due to CA’s free radical scavenging potential [26]. This may preserve cell membrane function including ion transport and membrane fluidity.
In the current study, the effect of chronic Al exposure on brain monoaminergic systems as stress response indicators was assayed. Primarily, the data presented in this work elicited a marked depletion in the brain monoaminergic system (5-HT, DA, E, NE and 5-HT) levels, which negatively correlated with the brain Al level. 5-HT is an excitatory neurotransmitter synthesized from tryptophan via the intermediate 5-hydroxytryptophane. The decreases in the brain 5-HT levels are associated with a decrease in the synthesis of 5-HT and loss of serotonergic neurons [40]. The decline in the levels of DA, E and NE neurotransmitters may be linked to their closely related synthetic route. These neurotransmitters are derived from tyrosine, which is converted to DA by the enzymes tyrosine hydroxylase and DOPA decarboxylase. DA is converted through the action of dopamine-β-hydroxylase to NE [41]. Similar decreases were previously reported by Fernandez-Davila et al., [42]. In the current study CA supplementation might be useful in maintaining brain neurotransmitters levels to some extent.
Al intoxication leads to marked astrocytes and microglial activation that contribute to the pathogenesis of neuroinflammation and neurodegeneration [43]. Chronic administration of AlCl3 causes a greater inflammatory response over immunoreactivity of astrocytes and microglia associated with cognitive dysfunction and disease [44]. Reactive gliosis and the associated proinflammatory cytokines/chemokines could be result of inflammation and neuronal injury in the rat brain [45]. Inhibition of pro-inflammatory mediators would be a better approach in regulating the progression of neurodegenerative disorders. The pro-inflammatory cytokines, TNF-α and iNOS, contribute to neuronal damage and death in vivo and in vitro [46]. Activation of iNOS in glial cells is a key step in neuroinflammation and is often related with TNF-α release. A potent proinflammatory cytokine TNF-α is synthesized by microglia, astrocytes and neurons [47]. TNF-α together with other transcription factors (NF-?B, nuclear factor kappa B) involved in cell responses including inflammation, proliferation, cell migration and apoptosis [48]. NO, the enzymatic product of iNOS, is associated with neuronal death by inhibiting mitochondrial respiration and excitotoxicity [49]. iNOS is normally not expressed in the brain, but is induced by pro-inflammatory cytokines and pathogenic components like endotoxins. To explore the mechanism of CA on the attenuation of AlCl3-induced neuroinflammation, the expression of TNF-α and iNOS was detected in the rat brain. Reduced expressions of TNF-α and iNOS were observed in the cortex of rats treated with CA, which demonstrates that CA exerts anti-inflammatory effect. Hence, we assume that CA regulates focal microglia activation and proinflammatory cytokine production in cortex and this antioxidant is an effective experimental tool to reduce the brain lesions associated with inflammatory responses [21]. It also influences memory performance and, hence, CA can be utilized as a protective agent against neurodegenerative disorders [26].
Apoptosis is one of the mechanisms contributing to neuronal loss in AlCl3 induced neurodegeneration. Aβ oligomers are majorly contributed to neuronal cell death by inducing apoptotic proteins during AlCl3 administration. Bcl-2 is a member of apoptotic regulator protein that plays a major role in the regulation of cell death under both the physiological as well as pathological conditions. Bcl-2 over expression is perceived as a survival factor to protect against neurons during brain injury [12,13]. In the present study, induction of AlCl3 resulted in a decrease in the activity of Bcl2 where as increased the activity of baxin cerebral cortex region of rat brain. The ratio of Bax/Bcl2 levels in cortex was turned in favour of apoptosis in AlCl3-administered rats that denotes that AlCl3 induces neuronal apoptosis in cortex region. Results of the present study are supported by documented report denoting the involvement of Bax, Bcl2 in AlCl3-induced apoptosis in rabbit [13,50]. Further, we demonstrate that treatment with CA reduces Bax/Bcl2 ratio thereby antagonizing apoptotic niche in cortex of AlCl3-administered rat brain. Mobilization of proapoptotic Bcl2 family signaling protein, Bax from cytosol to mitochondria causes mitochondrial outer membrane permeabilisation leading to cytochrome c expulsion from mitochondrial inter-membrane space leading to Caspase activation [51]. In this study, protein levels of cytochrome c and expressions of caspase-3 were significantly increased upon AlCl3-exposure in the cortex denotes that AlCl3-induces neuronal apoptosis. Results of present study are in accordance with documented reports denoting the involvement of caspase activation in many neurodegenerative disorders [52]. Treatment with CA attenuates AlCl3-induced cytochrome c expression and also its downstream effectors caspases-3. This indicates that CA exerts neuroprotective effect by inhibiting neuronal apoptosis and is further supported that dietary flavonoids are capable of inhibiting intrinsic apoptosis of neurons [53].
In summary, the present investigation sheds light on the potent neuroprotective potential of CA in AlCl3-induced neurotoxicity. Our observations suggest that CA helps to maintain brain neurotransmitters levels and further regulates apoptotic neuronal cell death, focal microglia activation and inflammation in the cortex. Therefore, it is sensible to suggest from the previous and the present studies that CA may be used as therapeutic agents in the treatment of cognitive dysfunction-associated disorders such as Alzheimer’s disease. Future research may be directed towards the use of CA in clinical trials to evaluate its neuroprotective effect on humans.
CONFLICT OF INTEREST
ACKNOWLEDGMENT
REFERENCES
- Yokel RA, McNamara PJ (2001) Aluminium toxicokinetics: an updated minireview. Pharmacol Toxicol 88: 159-167.
- Zaky A, Mohammad B, Moftah M, Kandeel KM, Bassiouny AR (2013) Apurinic/apyrimidinic endonuclease 1 is a key modulator of aluminum-induced neuroinflammation. BMC Neurosci 11: 14-26.
- Tomljenovic L (2011) Aluminum and Alzheimer's disease: after a century of controversy, is there a plausible link? J Alzheimers Dis 23: 567-598.
- Kawahara M, Kato-Negishi M (2011) Link between Aluminum and the Pathogenesis of Alzheimer's Disease: The Integration of the Aluminum and Amyloid Cascade Hypotheses. Int J Alzheimers Dis 2011: 276393.
- Andrasi E, Pali N, Molnár Z, Kosel S (2005) Brain aluminum, magnesium and phosphorus contents of control and Alzheimer-diseased patients. J Alzheimers Dis 4: 273-284.
- Platt B, Fiddler G, Riedel G, Henderson Z (2001) Aluminium toxicity in the rat brain: histochemical and immunocytochemical evidence. Brain Res Bull 55: 257-267.
- Exley C (2004) The pro-oxidant activity of aluminum. Free Radic Biol Med 36: 380-387.
- Lukiw WJ, Percy ME, Kruck TP (2005) Nanomolar aluminum induces pro-inflammatory and pro-apoptotic gene expression in human brain cells in primary culture. J Inorg Biochem 99: 1895-1898.
- Aremu DA, Meshitsuka S (2005) Accumulation of aluminum by primary cultured astrocytes from aluminum amino acid complex and its apoptotic effect. Brain Res 1031: 284-296.
- Mattson MP (2000) Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol 1: 120-129.
- Okouchi M, Ekshyyan O, Maracine M, Aw TY (2007) Neuronal apoptosis in neurodegeneration. Antioxid Redox Signal 9: 1059-1096.
- Zhang H, Zhang YW, Chen Y, Huang X, Zhou F, et al. (2012) Appoptosin is a novel pro-apoptotic protein and mediates cell death in neurodegeneration. J Neurosci 32: 15565-15576.
- Ghribi O, Herman MM, Spaulding NK, Savory J (2002) Lithium inhibits aluminum-induced apoptosis in rabbit hippocampus, by preventing cytochrome c translocation, Bcl-2 decrease, Bax elevation and caspase-3 activation. J Neurochem 82: 137-145.
- Ghribi O, Herman MM, Forbes MS, DeWitt DA, Savory J (2001) GDNF protects against aluminum-induced apoptosis in rabbits by upregulating Bcl-2 and Bcl-XL and inhibiting mitochondrial Bax translocation. Neurobiol Dis 8: 764-773.
- Mattson MP, Partin J, Begley JG (1998) Amyloid beta-peptide induces apoptosis-related events in synapses and dendrites. Brain Res 807: 167-176.
- Bernstein AI, Garrison SP, Zambetti GP, O'Malley KL (2011) 6-OHDA generated ROS induces DNA damage and p53- and PUMA-dependent cell death. Mol Neurodegener 6: 2.
- Hyman BT, Yuan J (2012) Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat Rev Neurosci 13: 395-406.
- Jayathirtha MG, Mishra SH (2004) Preliminary immunomodulatory activities of methanol extracts of Eclipta alba and Centella asiatica. Phytomedicine 11: 361-365.
- Zainol MK, Abd-Hamid A, Yusof S, Muse R (2003) Antioxidative activity and total phenolic compounds of leaf, root and petiole of four accessions of Centella Asiatica (L.) Urban. Food Chemistry 81: 575-581.
- Hussin M, Abdul-Hamid A, Mohamad S, Saari N, Ismail M, et al. (2007) Protective effect of Centella asiatica extract and powder on oxidative stress in rats. Food Chemistry 100: 535-541.
- Somchit MN, Sulaiman MR, Zuraini A, Samsuddin L, Somchit N, et al. (2004) Antinociceptive and antiinflammatory effects of Centella asiatica. Indian Journal of Pharmacology 36: 377-380.
- Babu TD, Kuttan G, Padikkala J (1995) Cytotoxic and anti-tumour properties of certain taxa of Umbelliferae with special reference to Centella Asiatica (L.) Urban. J Ethnopharmacol 48: 53-57.
- Soumyanath A, Zhong YP, Gold SA, Yu X, Koop DR, et al. (2005) Centella Asiatica accelerates nerve regeneration upon oral administration and contains multiple active fractions increasing neurite elongation in-vitro. J Pharm Pharmacol 57: 1221-1229.
- Appa Rao MVR, Srinivasan K, Koteswara Rao T (1977) The effect of Centella Asiatica on the general mental ability of mentally retarded children. Indian Journal of Psychiatry 19: 54-59.
- Dhanasekaran M, Holcomb LA, Hitt AR, Tharakan B, Porter JW, et al. (2009) Centella Asiatica extract selectively decreases amyloid beta levels in hippocampus of Alzheimer's disease animal model. Phytother Res 23: 14-19.
- Amjad S, Umesalma S (2015) Protective Effect of Centella Asiatica against Aluminium-Induced Neurotoxicity in Cerebral Cortex, Striatum, Hypothalamus and Hippocampus of Rat Brain- Histopathological, and Biochemical Approach. J Mol Biomark Diagn 6: 212.
- Llobet JM, Domingo JL, Gómez M, Tomás JM, Corbella J (1987) Acute toxicity studies of aluminium compounds: antidotal efficacy of several chelating agents. Pharmacol Toxicol 60: 280-283.
- Glowinski J, Iversen LL (1966) Regional studies of catecholamines in the rat brain. I. The disposition of [3H]norepinephrine, [3H]dopamine and [3H]dopa in various regions of the brain. J Neurochem 13: 655-669.
- Zumkley H, Bertram HP, Lison A, Knoll O, Losse H (1979) Aluminum, zinc and copper concentrations in plasma in chronic renal insufficiency. Clin Nephrol 12: 18-21.
- Byers JP, Masters K, Sarver JG, Hassoun EA (2006) Association between the levels of biogenic amines and superoxide anion production in brain regions of rats after subchronic exposure to TCDD. Toxicology 228: 291-298.
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265-275.
- Towbin H, Stahelin H, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76: 4350-4354.
- Heneka MT, O'Banion MK (2007) Inflammatory processes in Alzheimer's disease. J Neuroimmunol 184: 69-91.
- Hong J, Cho IH, Kwak KI, Suh EC, Seo J, et al. (2010) Microglial toll-like receptor 2 contributes to kainic acid induced glial activation and hippocampal neuronal cell death. J Biol Chem 50: 39447-39457.
- Bondy SC (2010) The neurotoxicity of environmental aluminum is still an issue. Neurotoxicology 31: 575-581.
- Ong WY, Farooqui T, Kokotos G, Farooqui AA (2015) Synthetic and natural inhibitors of phospholipases A2: their importance for understanding and treatment of neurological disorders. ACS Chem Neurosci 6: 814-831.
- Doknark S, Mingmalairak S, Vattanajun A, Tantisira B, Tantisira MH (2014) Study of ameliorating effects of ethanolic extract of Centella Asiatica on learning and memory deficit in animal models. J Med Assoc Thai 97: 68-76.
- Kawahara M (2005) Effects of aluminum on the nervous system and its possible link with neurodegenerative diseases. J Alzheimers Dis 8: 171-182.
- Yokel RA (2002) Aluminum chelation principles and recent advances. Coord Chem Rev 228: 97-113.
- Kumar S (2002) Aluminium-induced changes in the rat brain serotonin system. Food Chem Toxicol 40: 1875-1880.
- Mycek MJ, Harvey RA, Champe PC (Eds.) (2007) Farmacologia. 2nd ed. McGraw Hill, Mexico City pp. 65-82.
- Fernández-Dávila ML, Razo-Estrada AC, García-Medina S, Gómez-Oliván LM, Piñón-López MJ, et al. (2012) Aluminum-induced oxidative stress and neurotoxicity in grass carp (Cyprinidae--Ctenopharingodon idella). Ecotoxicol Environ Saf 76: 87-92.
- Kaushik DK, Mukhopadhyay R, Kumawat KL, Gupta M, Basu A (2012) Therapeutic targeting of Krüppel-like factor 4 abrogates microglial activation. J Neuroinflammation 9: 57.
- Walton JR (2012) Cognitive Deterioration and Associated Pathology Induced by Chronic Low-Level Aluminum Ingestion in a Translational Rat Model Provides an Explanation of Alzheimer's Disease, Tests for Susceptibility and Avenues for Treatment. Int J Alzheimers Dis Pg no: 17.
- O'Callaghan JP, Sriram K, Miller DB (2008) Defining "neuroinflammation". Ann N Y Acad Sci 1139: 318-330.
- Wang YP, Wu Y, Li LY, Zheng J, Liu RG, et al. (2011) Aspirin-triggered lipoxin A4 attenuates LPS-induced pro-inflammatory responses by inhibiting activation of NF-κB and MAPKs in BV-2 microglial cells. J Neuroinflammation 8: 95.
- Park KM, Bowers W J (2010) Tumor necrosis factor-alpha mediated signaling in neuronal homeostasis and dysfunction. Cell Signal 22: 977-983.
- Tan ZS, Beiser AS, Vasan RS, Roubenoff R, Dinarello CA,et al. (2007) Inflammatory markers and the risk of Alzheimer disease: The Framingham Study. Neurology 68: 1902-1908.
- Bal-Price A, Brown GC (2001) Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci 2: 6480-6491.
- Niu Q, Wang LP, Chen YL, Zhang HM (2005) [Relationship between apoptosis of rat hippocampus cells induced by aluminum and the copy of the bcl-2 as well as bax mRNA]. Wei Sheng Yan Jiu 34: 671-673.
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, et al. (1997) Cytochrome c and dATP-dependent formation of Apaf-1/Caspase-9 complex initiates an apoptotic protease cascade. Cell 91: 479-489.
- Perier C, Bové J, Wu DC, Dehay B, Choi DK, et al. (2007) Two molecular pathways initiate mitochondria-dependent dopaminergic neurodegeneration in experimental Parkinson's disease. Proc Natl Acad Sci U S A 104: 8161-8166.
- Prakash A, Kumar A (2013) Mitoprotective effect of Centella Asiatica against aluminum-induced neurotoxicity in rats: possible relevance to its anti-oxidant and anti-apoptosis mechanism. Neurol Sci 34: 1403-1409.
Citation: Amjad S, Umesalma S (2018) Centella Asiatica Extracts Regulates Aluminium Chloride-Induced Neurotoxicity in Rats: Impact on Inflammation, Apoptosis and Biogenic Amine Levels. J Pharmacol Pharmaceut Pharmacovigil 2: 007.
Copyright: © 2018 Shaik Amjad, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.