Interactions between Parasite and Host in Human Visceral Leishmaniasis
*Corresponding Author(s):
Harizanov RDepartment Of Parasitology And Tropical Medicine, National Center Of Infectious And Parasitic Diseases, Sofia, Bulgaria
Tel:+35929446999,
Fax:+35928438002
Email:harizanov@ncipd.org
Abstract
Leishmaniasis is one of the most important Neglected Tropical Diseases (NTDs) affecting mainly the poorest social groups in many countries of the world. Visceral form of the disease is caused by protozoa belonging to Leishmania donovani complex. The clinical manifestations arise from a number of complex interactions between cells of the causative agent and the host. Our aim is briefly to introduce some of the pathogenetic mechanisms in visceral leishmaniasis with a belief that detailed study and understanding of the disease pathology could lead to more effective methods for rapid diagnosis and effective treatment.
Keywords
DISEASE
Visceral leishmaniasis is a protozoan, vector born disease characterized by a chronic course, undulating fever, splenic and hepatic enlargement and anemia to complete secondary pancytopenia and immunosuppression.
HISTORY
In 1900 William Leishman performed pathological examination of the body of a British soldier who died from dum-dum fever (Dum Dum is an area in north Kolkata, formerly known as Calcutta). The deceased suffered from febrile episodes, anemia, muscle atrophy and enlargement of the spleen. Examining spleen specimens Leishman discovered a huge number of oval bodies measuring 2-3 µm. He published his findings in British Medical Journal on 11 May 1903. A month later, similar finding in a material from enlarged spleen of a patient who died of suspected malaria in Madras (India), described Charles Donovan. Laveran and Mesnil classified this parasite as protozoa. Subsequently, Ronald Ross, using Leishmans staining technique named these protozoan microorganisms "Leishman bodies", and Manson named them "Leishman-Donovan bodies"-in memory of the two English scientists. The name, "kala-azar" (black disease) was given by McNaught, who described the disease in India. Swaminath et al., using volunteers proved that the disease is transmitted to humans by insects and found those specific biological carriers are bloodsucking insects from genus Phlebotomus. For first time Rogers was able to cultivate the parasite in citrated blood, but his attempts to maintain the strain by passaging were unsuccessful. Later, Ch. Nicolle was able to cultivate and maintain Leishmania strain by passaging and also to induce experimental disease in dogs and monkeys. He first highlights the epidemiological link between man and dog [1,2].
ETIOLOGY
Taxonomy
Genus Leishmania belongs systematically to Kingdom Protista; Subkingdom Protozoa; Family Tripanosomatidae [6]. Taxonomic identification of the different species Leishmania is based on two types of features-external (clinical manifestations, geographical distribution, epidemiological cycle) and internal (morphological and molecular structure) [4,7].
Parasite classification at genus level has been based on global taxonomics derived in the 1990s using isoenzyme technique in comparison with reference strains. Identification depends on the employed method, e.g. zymodemes (parasite populations with common isoenzyme patterns identified electrophoretically) or schizodemes (parasite populations defined by shared 'fingerprint patterns' obtained by a process involving digestion of kinetoplast DNA by restriction enzymes). The results are relevant in descriptive epidemiology and allows grouping of the parasites into hierarchies that suggest their evolutionary relations [8].
In the Mediterranean basin, L infantum displays a broad enzymatic polymorphism with 20 different zymodemes, of which 18 have been found in humans. L. infantum MON-1 is the most frequent zymodeme in humans and is usually responsible for VL along the Mediterranean, region more rarely for CL [9].
Morphology and life cycle
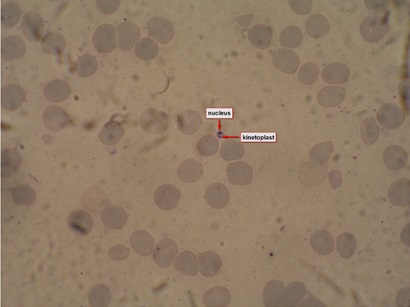 Figure 1: Leishmanial amastigote in bone marrow specimen from a patient with visceral leishmaniasis.
Figure 1: Leishmanial amastigote in bone marrow specimen from a patient with visceral leishmaniasis.
When taking a blood meal from human or animal carrier the female sand fly ingest with the blood also hosts cells invaded by amastigotes. Then the amastigotes are released into the midgut of the digestive tract of the insect where are transformed into elongated flagellate promastigotes (procyclic promastigotes) and in a few days later their number increases significantly. The promastigotes migrate to the front section of the digestive tract where are transformed into short, spherical, non-dividing promastigotes (metacyclic promastigotes) and when the phlebotomine sand fly takes another blood meal it will inoculate 10 to 200 promastigotes (Figure 2) in the dermis of a vertebrate host [8,10,11]. From there they fall into the phagocytic cells of the immune system, including macrophages, dendritic cells and neutrophils where will transform again to amastigotes and begin cycle of reproduction [14]. The infected macrophages spread from the dermis to other tissues and organs such as spleen, liver and bone marrow. Different species Leishmania possess specific tropism, and some of them are dermatotropic (causing cutaneous and mucocutaneous leishmaniasis), while others are viscerotropic (causing visceral leishmaniasis). The reasons for existence of specific tropism are not clear yet, but it is assumed that depends on some genetic features of the host and the parasite, as well as some external factors such as temperature. For example dermatotropic Leishmania spp. can not realize their biological cycle at the ambient temperature of the internal organs of a vertebrate host as are able the viscerotropic Leishmania spp.
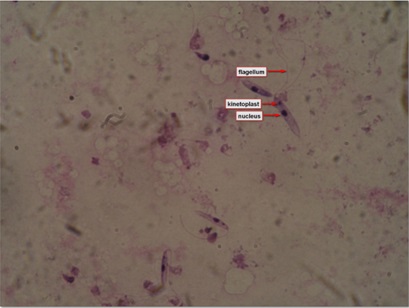 Figure 2: Leishmanial promastigotes from culture in triple N (NNN-Novy, MacNeal, Nicolle) medium.
Figure 2: Leishmanial promastigotes from culture in triple N (NNN-Novy, MacNeal, Nicolle) medium.EPIDEMIOLOGY
Geographic distribution
In South-East Asia visceral leishmaniasis is the main form of the disease. Transmission most often occurs in rural areas below 600m above sea level, with a heavy annual rainfall, mean humidity above 70%, a temperature range of 15-38°C, abundant vegetation, subsoil water and alluvial soil. The disease is most common in agricultural villages where houses are frequently constructed with mud walls and earthen floors, and livestock is living close to humans [4,8,27].
In East Africa there are frequent outbreaks of visceral leishmaniasis in in the northern Acacia-Balanite savanna and the southern savanna and forest areas where sandflies live around termite mounds [4,8,27].
Americas-visceral leishmaniasis is very similar to that found in the Mediterranean Basin [4,8,27].
The disease is highly endemic in the Indian subcontinent and in East Africa. An estimated 200, 000 to 400, 000 new cases of VL occur worldwide each year. Over 90% of new cases occur in six countries: Bangladesh, Brazil, Ethiopia, India, South Sudan, and Sudan [27].
PATHOGENESIS AND PATHOLOGY
Pathogenesis and histological changes in individuals with uncompromised immunity
Macrophages, the primary target of intracellular Leishmania infection, may take on distinct phenotypes in response to parasite signals and inflammatory stimuli within the infected microenvironment. Classically activated (M1) macrophages respond to IFN? and microbial products by generating antimicrobial molecules that effectively kill Leishmania and other intracellular pathogens. Central to the killing of intracellular parasites is the production of nitric oxide by the action of inducible nitric oxide synthase 2 on the substrate L-arginine. In contrast, alternatively activated or M2 macrophages, which are typically generated by exposure to type 2 cytokines (IL-4, IL-13), fail to produce antimicrobial effector molecules to kill intracellular pathogens and serve to dampen inflammation and promote wound healing [33,34]. However, as other infectious agents, most Leishmania species have evolved effective strategies to evade the innate immune response during the early moments of infection, and this is by rapidly blocking the induction and regulation of key host cell functions including production of nitric oxide, tumour necrosis factor-alpha, interleukin-12, and radical oxygen species [35]. Peripheral blood mononuclear cells from patients with manifested VL typically do not proliferate or produce IFNγ in response to Leishmanial Antigen (LAg). A few months after completion of therapy, following cure, proliferative and cytokine responses to LAg are usually detectable [36]. Leishmania parasites have been shown to alter host-cell signaling pathways to promote their survival. Activation of host Src homology 2 domain containing tyrosine phosphatases by Leishmania infection results in global dephosphorylation of tyrosine residues, leading to deactivation of a variety of signaling pathways including JAK/STAT, NF-?B, IRF-1, and MAP kinases. Increased concentrations of secondary messengers including Calcium (Ca2+), inositol lipids, inositol phosphatases, and protein kinase C have also been observed following Leishmania infection. Induction of the negative regulatory proteins, suppressors of cytokine signaling, have been characterized following Leishmania infection and interfere with cytokine signaling and host cell activation [12].
On the sites of inoculations are formed small granulomas (leishmaniomas), which consists of histiocytes containing parasites. They are surrounded by epithelioid cells, which are gradually transformed into giant cells. Subsequent development depends on the host immune response, ranging from missing to a response that leads to a full eradication of the parasite. From the site of inoculation, and after transformation into amastigotes the leishmania parasites localize in the regional lymph nodes, from where they pass into the blood stream and through it to other cells of the reticuloendothelial system of the end host. It develops hyperplasia of the reticuloendothelial cells of the liver, spleen, bone marrow, lymph nodes, lining of the small intestine, and other lymphoid tissues. Increase of the spleen size is due to reticuloendothelial proliferation and, in some cases can be so extreme that its caudal part may reach the iliac fossa. The spleen is with dense consistency and full with congested blood. In the acute stage of the disease its capsule is smooth, within the pulp are observed multiple infarct areas and massive intracellular invasion of the histiocytes by amastigotes. Histologic studies of liver changes in visceral leishmaniasis showed that the Kupffer cells in the portal areas contain numerous parasites [4,8,10,16,20,24]. In most cases, efficient immune responses to L. donovani in the liver depend on the formation of granulomas, a process influenced by chemokine production, subsequent recruitment of monocytes, neutrophils, CD4+ T cells and CD8+ T cells, production of inflammatory cytokines and activation of infected cells [37]. Lymph nodes may be enlarged and contain histiocytes infected with amastigotes. Bone marrow is poor in cellular elements as myelocytes and promyelocytes predominate. At the beginning hematopoiesis is normal, but in the course of illness the life of erythrocites and leukocytes shortens and that leads to anemia and granulocytopenia with relative lymphoid and monocytosis. Subsequently is reduced the synthesis of prothrombin. This in combination with growing thrombocytopenia can lead to severe bleeding from mucous membranes. The course of the disease may be complicated with diarrhea, ulceration of the intestinal mucosa, and enteritis in untreated cases. In the late stage may develop secondary infections such as pneumonia and tuberculosis, which often lead to death. Activation of the complement system may enhance developing anemia due to circulating immune complexes, but complications such as development of glomerulonephritis have been reported rarely. Reticuloendothelial massive proliferation induces enlargement of the liver and spleen, which upon proper treatment may restore their original size [4,8,20,24,38,39].
Pathogenesis in leishmania/HIV co-infection
IMMUNITY
T-cells immune response in visceral leishmaniasis
B-cells immune response in visceral leishmaniasis
Target of the humoral immune response are some heat shock proteins (Hsp). Antibodies against Hsp 70 have been detected in 92% of patients with VL [44]. In blood sera of the patients can be found circulating immune complexes with immunoglobulins of classes A, G and M. Skin patch delayed hypersensitivity reaction is suppressed or even absent. After recovery this sensitivity is restored [40,52].
CLINICAL PRESENTATION AND COMPLICATIONS
Visceral leishmaniasis is a kind of reticuloendotheliosis and is directly related to the immune status of infected individuals. Clinical manifestations of VL can range from asymptomatic subclinical carrier to manifested, severe forms of the disease. After incubation period ranging from 10 days to more than a year (mean of 2 and 4 months) appear the first symptoms of the disease. Depending on the species of viscerotropic leishmaniae the beginning may be indolent or noticeable-protracted or severe. In the first case the symptoms can be vague, with a sense of discomfort, while acute onset disease resembles typhoid fever or malaria. Described are several clinical forms of VL [24].
Light subclinical (oligosymptomatic) form of VL
Classical form of VL
Initial stage: The earliest clinical symptom is development of primary lesion-ulcer at the site of inoculation, which can be detected in the prodromal period prior general clinical manifestations to appear. Typically, in the initial stage the first manifestation of VL is increased body temperature, which is constant or remitting, and later can become intermittent with two or three peaks a day [20,24,57-60].
Stage of splenohepatomegaly: Gradually, amid prolonged febrile condition develops enlargement of the spleen and liver, lymphadenopathy, accompanied in some cases by acute abdominal pain. Darkening of the skin of the face, hands, feet and abdomen is observed in the Indian version of VL (kala-azar). Clinical examination reveals that splenic enlargement is more pronounced than hepatomegaly. The spleen is usually palpable at about 5-15 cm below the left rib arc. According to some authors, splenomegaly may be absent in about 5% of the cases [61]. Anemia, weight loss and pronounced splenohepatomegaliy occur with disease progression. Anemia is a symptom, almost always accompanying visceral leishmaniasis, and in some cases may be of high grade. It is usually normocytic and normochromic, and is caused by various factors, including bone marrow damage by the parasites, signs of hypersplenism, haemorrhage and hemolysis. Leukopenia is common, and in some cases on this background develop secondary infections. Thrombocytopenia may lead to bleeding (haemorrhagic) diathesis [57,58]. In some cases of VL can be found hypergammaglobulinemia, circulating immune complexes and positive rheumatoid factor. Seldom, the circulating immune complexes can cause development of glomerulonephritis in the kidneys [62]. Also have been described cases of combination of VL with acute hepatitis [63], bacteremia [64], Guillain-Barre syndrome [65]. Malabsorption is a significant problem in children with VL and is a consequence of adverse effects of the cell-mediated immunity [66]. In cases of disease in India and Sudan have been observed Post-kala-azar Dermal Leishmaniasis (PKDL). These nodular or papular skin lesions contain large number of amastigotes and are of great epidemiological importance for the spread of VL.
Stage of cachexia: Usually develops because of delayed diagnosis and/or improper treatment. In patients in this stage is observed a significant reduction of body weight. This is accompanied by aggravation of the haematological changes with development of hemorrhagic diathesis. Without treatment, the clinically manifested VL is usually fatal [8,20,57,58,67].
Sudanese clinical variation of VL (Sudanese disease; Killing disease)
VL associated with AIDS
Diagnosis and Treatment options
In most cases visceral leishmaniasis is curable disease. Several treatment options are available but their usage and efficacy depend on a variety of factors. These factors include parasite species, severity of the symptoms and the immune status of the host. The only treatment currently available for visceral leishmaniasis relies on chemotherapy [12,77]. Classical, first-line drug treatments include pentavalent antimonials compounds, stibogluconate (Pentostam) or meglumine antimonite (Glucantime). Pentavalent antimonials have been in use for more than 50 years and are the recommended treatment by the World Health Organization (WHO) [4,8]. Other common drugs include amphoterecin B, lipid formulations of amphoterecin B, including liposomal amphotericin B, amphotericin B lipid complex and amphotericin B colloidal dispersion, pentamidine, miltefosine (Impavido or Miltex) and imidazoles, allopurinol (Zyloric), or paromomycin among others.
However, several obstacles remain in the treatment of visceral leishmaniasis Most treatments require extensive (weeks to months) and invasive (intramuscular or intravenous) modes of administration and there is a high rate of significant drug-related toxic side effects. The cost of treatment is often well beyond the means of many patients and severe toxicity may require secondary treatment, adding to the already high cost of therapy [12,69].
CONCLUSION
Host-parasite interactions are many and various in nature. In the review we have tried to present briefly some interactions between the agents of visceral leishmaniasis and hosts cells. Their further study could lead to improved diagnostic methods and development of new therapeutic formulations and vaccines which to introduce in the clinical practice.
REFERENCES
- Thakur CP, History of Kala-azar.
- Gibson ME (1983) The identification of kala-azar and the discovery of Leishmania donovani. Med Hist 27: 203-213.
- Lainson R, Shaw JJ (1987) Evolution, classification and geographical distribution. The Leishmaniasis in Biology and Medicine 1: 1-120.
- Control of the leishmaniasis (1990) WHO, Technical Report Series. Geneva, Switzerland.
- Shaw JJ (1994) Taxonomy of the genus Leishmania: present and future trends and their implications. Mem Inst Oswaldo Cruz 89: 471-478.
- Golemanski V, Schischinyova M (2001) Zoology of Invertebrates. Gera Art, Sofia.
- Evans TG (1993) Leishmaniasis. Infect Dis Clin North Am 7: 527-546.
- Control of the leishmaniasis (2010) Report of a meeting of the WHO Expert Committee on the Control of Leishmaniases, Geneva.
- Sulahian A, Garin YJF, Pratlong F, Dedet JP, Derouin F (1997) Experimental pathogenicity of viscerotropic and dermotropic isolates of Leishmania infantum from immunocompromised and immunocompetent patients in a murine model. FEMS Immunology and Medical Microbiology 17: 131-138.
- Neva FA, Brown HW (1994) Basic clinical parasitology. (6thedn). Prentice-Hall International Inc.
- Hommel M (1999) Visceral leishmaniasis: Biology of the parasite. J Infect 39: 101-111.
- Boggiatto P (2010) Leishmaniasis: immunologic indicators of clinical progression and mechanisms of immune modulation. Graduate Theses and Dissertations. Iowa State University Paper.
- Bogitsh BJ, Carter CE, Oeltmann TN (2005) Human Parasitology. (3rdedn). Elsevier Academic Press.
- Sacks D, Sher A (2002) Evasion of innate immunity by parasitic protozoa. Nat Immunol 3: 1041-1047.
- McCall LI, Zhang WW, Matlashewski G (2013) Determinants for the development of visceral leishmaniasis disease. PLoS Pathog 9: 1003053.
- Scott P (1985) Impaired macrophage leishmanicidal activity at cutaneous temperature. Parasite Immunol 7: 277-288.
- Wittner M, Tanowitz HB (2000) Leishmaniasis in infants and children. Semin Pediatr Infect Dis 11: 196–201.
- Callahan HL, Portal IF, Bensinger SJ, Grogl M (1996) Leishmania spp: temperature sensitivity of promastigotes in vitro as a model for tropism in vivo. Exp Parasitol 84: 400-409.
- McCall LI, Matlashewski G (2010) Localization and induction of the A2 virulence factor in Leishmania: evidence that A2 is a stress response protein. Mol Microbiol 77: 518-530.
- Garcia LS, Buckner DA (1993) Diagnostic Medical Parasitology. (2ndedn). American Society for Microbiology, Washington, USA.
- Seaman J, Mercer AJ, Sondorp E (1996) The epidemic of visceral leishmaniasis in western Upper Nile, southern Sudan: course and impact from 1984 to 1994. Int J Epidemiol 25: 862-871.
- Alvar J, Cañavate C, Gutiérrez-Solar B, Jiménez M, Laguna F, et al. (1997) Leishmania and human immunodeficiency virus coinfection: the first 10 years. Clin Microbiol Rev 10: 298-319.
- Desjeux P (1996) Leishmaniasis. Public health aspects and control. Clin Dermatol 14: 417-423.
- Berman JD (1997) Human leishmaniasis: clinical, diagnostic, and chemotherapeutic developments in the last 10 years. Clin Infect Dis 24: 684-703.
- Guerin PJ, Olliaro P, Sundar S, Boelaert M, Croft SL, et al. (2002) Visceral leishmaniasis: current status of control, diagnosis, and treatment, and a proposed research and development agenda. Lancet Infect Dis 2: 494-501.
- Harizanov R, Rainova I, Tzvetkova N, Kaftandjiev I, Bikov I, et al. (2013) Geographical distribution and epidemiological characteristics of visceral leishmaniasis in Bulgaria, 1988 to 2012. Euro Surveill 18: 20531.
- Leishmaniasis (2014) WHO, Fact sheet N°375.
- Jefferies D, Livesey JL, Molyneux DH (1986) Fluid mechanics of bloodmeal uptake by Leishmania-infected sandflies. Acta Trop 43: 43-53.
- Killick-Kendrick R (1986) The transmission of leishmaniasis by the bite of the sandfly J Roy Army Med Corps 132: 134-140.
- Warburg A, Schlein Y (1986) The effect of post-bloodmeal nutrition of Phlebotomus papatasi on the transmission of Leishmania major. Am J Trop Med Hyg 35: 926-930.
- Schlein Y, Jacobson RL, Messer G (1992) Leishmania infections damage the feeding mechanism of the sandfly vector and implement parasite transmission by bite. Proc Natl Acad Sci USA 89: 9944-9948.
- Bates PA (2007) Transmission of Leishmania metacyclic promastigotes by phlebotomine sand flies. Int J Parasitol 37: 1097-1106.
- Martinez FO, Helming L, Gordon S (2009) Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol 27: 451-483.
- Osorio EY, Travi BL, da Cruz AM, Saldarriaga OA, Medina AA, et al. (2014) Growth Factor and Th2 Cytokine Signaling Pathways Converge at STAT6 to Promote arginase expression in progressive experimental visceral leishmaniasis. PLoS Pathog 10: 1004165.
- Olivier M, Atayde VD, Isnard A, Hassani K, Shio MT (2012) Leishmania virulence factors: focus on the metalloprotease GP63. Microbes Infect 14: 1377-1389.
- Kumar R, Nylén S (2012) Immunobiology of visceral leishmaniasis. Front Immunol 3: 251.
- Stanley AC, Engwerda CR (2007) Balancing immunity and pathology in visceral leishmaniasis. Immunol Cell Biol 85: 138-147.
- Gutierrez Y, Maksem JA, Reiner NE (1984) Pathologic changes in murine leishmaniasis (Leishmania donovani) with special reference to the dynamics of granuloma formation in the liver. Am J Pathol 114: 222-230.
- Giunchetti RC, Mayrink W, Carneiro CM, Corrêa-Oliveira R, Martins-Filho OA, et al. (2008) Histopathological and immunohistochemical investigations of the hepatic compartment associated with parasitism and serum biochemical changes in canine visceral leishmaniasis. Res Vet Sci 84: 269-277.
- Melhorn H (2004) Encyclopedic Reference of Parasitology. (2ndedn). Springer-Verlag Berlin Heidelberg, Newyork.
- Murray HW, Berman JD, Davies CR, Saravia NG (2005) Advances in leishmaniasis. Lancet 366: 1561-1577.
- Report of the Fifth Consultative Meeting on Leishmania/HIV Coinfection (2007) WHO, Addis Ababa, Ethiopia.
- Paredes R, Munoz J, Diaz I, Domingo P, Gurgui M, et al. (2003) Leishmaniasis in HIV infection. J Postgrad Med 49: 39-49.
- Wallace GR, Ball AE, MacFarlane J, el Safi SH, Miles MA, et al. (1992) Mapping of a visceral leishmaniasis-specific immunodominant B-cell epitope of Leishmania donovani Hsp70. Infect Immun 60: 2688-2693.
- Galvão-Castro B, Sá Ferreira JA, Marzochi KF, Marzochi MC, Coutinho SG, et al. (1984) Polyclonal B cell activation, circulating immune complexes and autoimmunity in human american visceral leishmaniasis. Clin Exp Immunol 56: 58-66.
- Sacks D, Sher A (2002) Evasion of innate immunity by parasitic protozoa. Nat Immunol 3: 1041-1047.
- Tse MT, Kwan P (2013) mTOR and its Physiological Impacts-Part II: Immunological Impact. Journal of Biochemical and Pharmacological Research 1:138-142.
- Contreras I, Gómez MA, Nguyen O, Shio MT, McMaster RW, et al. (2010) Leishmania-induced inactivation of the macrophage transcription factor AP-1 is mediated by the parasite metalloprotease GP63. PLoS Pathog 6: 1001148.
- Awasthi A, Mathur RK, Saha B (2004) Immune response to Leishmania infection. Indian J Med Res 119: 238-258.
- Sundar S, Reed SG, Sharma S, Mehrotra A, Murray HW (1997) Circulating T helper 1 (Th1) cell- and Th2 cell-associated cytokines in Indian patients with visceral leishmaniasis. Am J Trop Med Hyg 56: 522-525.
- Goto H, Prianti Md (2009) Immunoactivation and immunopathogeny during active visceral leishmaniasis. Rev Inst Med Trop Sao Paulo 51: 241-246.
- Pearson RD, Cox G, Jeronimo SM, Castracane J, Drew JS, et al. (1992) Visceral leishmaniasis: a model for infection-induced cachexia. Am J Trop Med Hyg 47: 8-15.
- Magill AJ, Grogl M, Johnson SC, Gasser RA Jr (1994) Visceral infection due to Leishmania tropica in a veteran of Operation Desert Storm who presented 2 years after leaving Saudi Arabia. Clin Infect Dis 19: 805-806.
- Badaró R, Jones TC, Lorenço R, Cerf BJ, Sampaio D, et al. (1986) A prospective study of visceral leishmaniasis in an endemic area of Brazil. J Infect Dis 154: 639-649.
- Badaro R, Jones TC, Carvalho EM, Sampaio D, Reed SG, et al. (1986) New perspectives on a subclinical form of visceral leishmaniasis. J Infect Dis 154: 1003-1011.
- Badaró R, Benson D, Eulálio MC, Freire M, Cunha S, et al. (1996) rK39: a cloned antigen of Leishmania chagasi that predicts active visceral leishmaniasis. J Infect Dis 173: 758-761.
- Genov G (1998) Practical Parasitology, (1stedn). Knowledge, Bulgarian.
- Petrov P (1992) Parasitology (1stedn), Medicine and Physical Education, Bulgarian.
- Conjivaram V (2013) Pediatric Leishmaniasis.
- Harizanov RN, Kaftandjiev IT, Jordanova DP, Marinova IB, Tsvetkova ND (2013) Clinical features, diagnostic tools, and treatment regimens for visceral leishmaniasis in Bulgaria. Pathog Glob Health 107: 260-266.
- Hashim FA, Ali MS, Satti M, el-Hassan AM, Ghalib HW, et al. (1994) An outbreak of acute kala-azar in a nomadic tribe in western Sudan: features of the disease in a previously non-immune population. Trans R Soc Trop Med Hyg 88: 431-432.
- Dutra M, Martinelli R, de Carvalho EM, Rodrigues LE, Brito E, et al. (1985) Renal involvement in visceral leishmaniasis. Am J Kidney Dis 6: 22-27.
- Hervás JA, Albertí P, Ferragut J, Canet R (1991) Acute hepatitis as a presenting manifestation of kala-azar. Pediatr Infect Dis J 10: 409-410.
- Garcés JM, Tomás S, Rubiés-Prat J, Gimeno JL, Drobnic L (1990) Bacterial infection as a presenting manifestation of visceral leishmaniasis. Rev Infect Dis 12: 518-519.
- Fasanaro AM, Scoleri G, Pizza V, Gaeta GB, Fasanaro A (1991) Guillain-Barré syndrome as presenting manifestation of visceral leishmaniasis. Lancet 338: 1142.
- Pearson RD, Sousa AQ (1996) Clinical spectrum of Leishmaniasis. Clin Infect Dis 22: 1-13.
- Sundar S, Rai M (2002) Laboratory diagnosis of visceral leishmaniasis. Clin Diagn Lab Immunol 9: 951-958.
- M Nail A, M Imam A (2013) Visceral leishmaniasis: Clinical and demographic features in an African population. Pak J Med Sci 29: 485-489.
- Chappuis F, Sundar S, Hailu A, Ghalib H, Rijal S, et al. (2007) Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nat Rev Microbiol 5: 873-882.
- Mukhopadhyay D, Dalton JE, Kaye PM, Chatterjee M (2014) Post kala-azar dermal leishmaniasis: an unresolved mystery. Trends Parasitol 30: 65-74.
- Montalban C, Calleja JL, Erice A, Laguna F, Clotet B, et al. (1990) Visceral leishmaniasis in patients infected with Human Immunodeficiency Virus (HIV). Co-operative Group for the Study of Leishmaniasis in AIDS. J Infect 21: 261-270.
- del Mar Sanz M, Rubio R, Casillas A, Guijarro C, Costa JR, et al. (1991) Visceral leishmaniasis in HIV-infected patients. AIDS 5: 1272-1274.
- Berenguer J, Moreno S, Cercenado E, Bernaldo de Quirós JC, García de la Fuente A, et al. (1989) Visceral leishmaniasis in patients infected with Human Immunodeficiency Virus (HIV). Ann Intern Med 111: 129-132.
- Rosenthal E, Marty P, Poizot-Martin I, Reynes J, Pratlong F, et al. (1995) Visceral leishmaniasis and HIV-1 co-infection in southern France. Trans R Soc Trop Med Hyg 89: 159-162.
- Pintado V, Martín-Rabadán P, Rivera ML, Moreno S, Bouza E (2001) Visceral leishmaniasis in Human Immunodeficiency Virus (HIV)-infected and non-HIV-infected patients. A comparative study. Medicine (Baltimore) 80: 54-73.
- Rosenthal E, Marty P, del Giudice P, Pradier C, Ceppi C, et al. (2000) HIV and Leishmania coinfection: a review of 91 cases with focus on atypical locations of Leishmania. Clin Infect Dis 31: 1093-1095.
- Frézard F, Demicheli C, Ribeiro RR (2009) Pentavalent antimonials: new perspectives for old drugs. Molecules 14: 2317-2336.
Citation: Harizanov, Rumen N, Kaftandjiev Iskren T (2014) Interactions between Parasite and Host in Human Visceral Leishmaniasis. J Cytol Tissue Biol 1: 001.
Copyright: © 2014 Harizanov R, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.