A 38 Year-old Man with Mediastinal Synovial Sarcoma: Case Report and Review of Literature
*Corresponding Author(s):
Anna EstivalDepartment Of Medical Oncology, Catalan Institute Of Oncology Badalona, Hospital Universitari Germans Trias I Pujol, Barcelona, Spain
Tel:+34 615445388,
Email:aestival@iconcologia.net
Abstract
Synovial Sarcoma (SS) is an infrequent sarcoma subtype usually located in limbs. Some cases have been described in uncommon regions, such as mediastinum. Mediastinal SS represents a diagnostic and therapeutic challenge.
Here, we present the case of a young patient admitted to the emergency room complaining of non-specific chest pain during the last three months. Imaging tests showed a huge mediastinal mass with right lung parenchyma and atrial septum infiltration. The final diagnose was an unresectable mediastinal SS.
Chemotherapy treatment with ifosfamide 2,000mg/m2 and doxorrubicin 20mg/m2 for 3 days every 21 days was initiated with neoadjuvant intention but the disease progressed after three cycles, so surgery was finally rejected. After two subsequent lines of treatment, the patient died due to congestive heart failure due to pericardium infiltration by SS, seven months after the initial diagnosis.
INTRODUCTION
Soft Tissue Sarcomas (STS) are rare tumors, representing less than 1% of all solid tumors diagnosed in the United States, and include subgroups with different features [1].
Synovial Sarcoma (SS) is an infrequent subtype (accounting for less than 10% of the sarcomas) with a very aggressive behavior and a poor prognosis at short term [2].
The pathological review shows immune reaction for epithelial markers and vimentin. Ninety percent of cases presents with SS18 (SYT) rearrangement on chromosome 18q11 [3,4]. SS typically occurs in young adults, with a 58% of patients diagnosed between 10 and 40 years, and with a median age at diagnosis of 35 years. The incidence does not differ according to gender [5-7].
The most common location is the lower limbs (70-80%) and predominantly affecting the juxtarticular region. Moreover, more than 8% of cases are found in the trunk, 7% in retroperitoneal region and 5% in head and neck [7,8]. Conversely to other sarcomas, SS presents with lymph node involvement more frequently (10-12% versus 3-5% in the rest of sarcomas). The lungs are the most frequent site of metastasis, and its spread in the liver is rare [9].
Some cases of SS with rare organ involvement have been reported such as mediastinal sarcomas. In these reports, the primary origin of the SS seems to be either pericardium or myocardium; however it is difficult to establish the real primary site due to the bulky nature of these tumors [10-17]. In addition, this unusual location contributes to aggravate the prognosis since the diagnosis of oligo symptomatic patients is frequently delayed, and the anatomical location represents a challenge for radical surgery. Consequently, a multidisciplinary approach is mandatory in these cases.
Here, we report a new case of a patient diagnosed with a mediastinal SS.
CASE PRESENTATION
A 38 year-old Caucasian male with neither prior medical history nor toxic habits presented with fever and productive cough. He also complained of non-specific chest pain during the last three months. A Chest-X-Ray was performed and it showed a right lower lobe consolidation with a mediastinal widening. Computed Tomography (CT) scan revealed a 112x112x105 cm mediastinal mass involving the right hilum, as well as subcarinal enlarged lymph nodes (Figure 1). A Magnetic Resonance Imaging (MRI) showed pleural and right lung parenchyma infiltration and a collapsed right atrium (50%) with atrial septum invasion in relation to the mediastinal mass (Figure 2).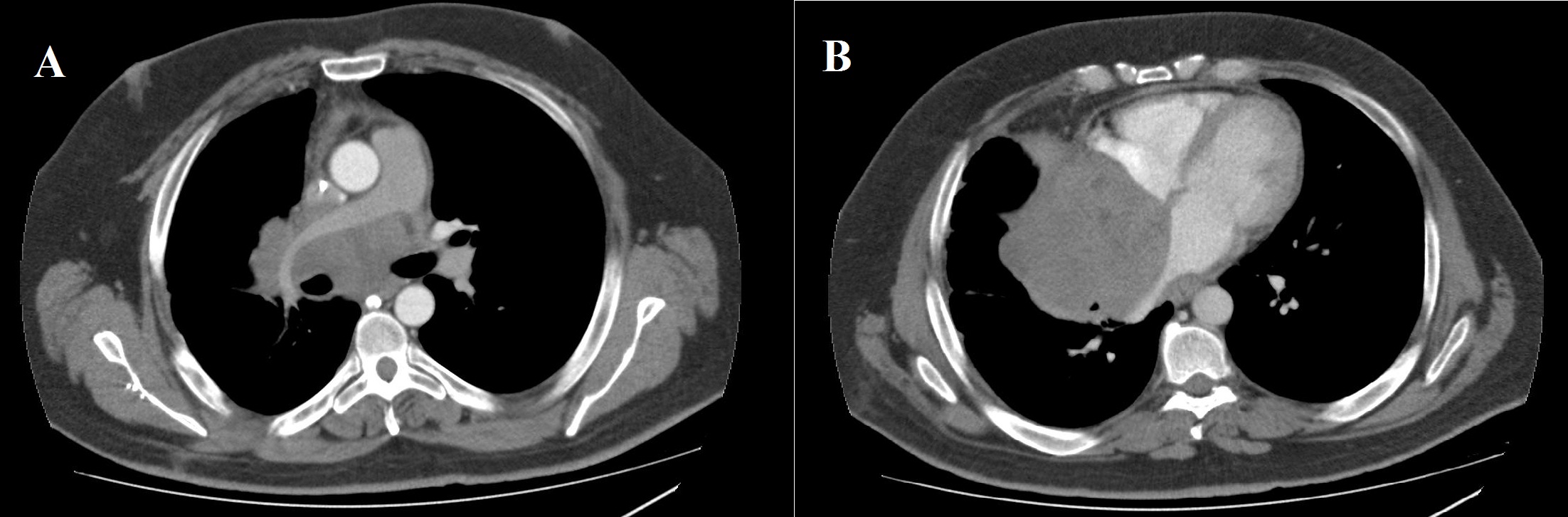
Figure 1: Computed tomography scan (mediastinal window). a) Axial images showing an 11 cm mediastinal mass involving the right pulmonary artery; b) Right atrium, and atrial septum.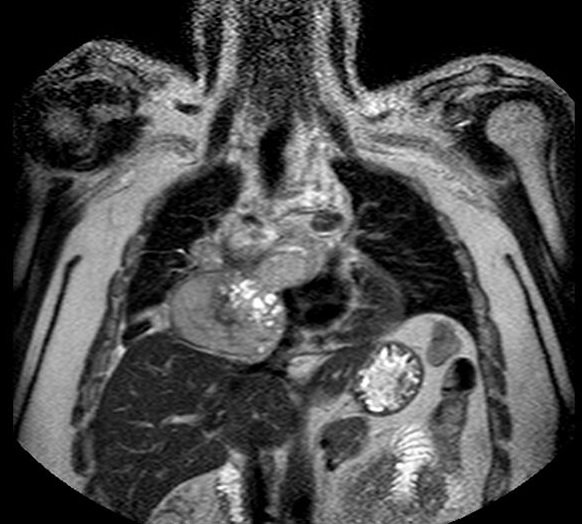
Figure 2: Magnetic resonance imaging. Coronal image showing pleural and right lung parenchyma infiltration and a collapsed right atrium with atrial septum invasion in relation to the tumour.
A bronchoscopy did not detect abnormalities. Patient underwent a mediastinoscopy that did not provide sufficient diagnostic material. Finally, a right thoracotomy was performed. The histological sample showed elongated cells with fine chromatin, scarce cytoplasm and occasional nucleolus. The cytoplasm had moderate amount of glycogen and some mitochondria. The cytoplasmatic membrane showed abundant filopodi. The cells were joined by desmosoma and rudimentary cell junctions (Figure 3a). Immunohistochemistry (IHC) showed positivity for Epithelial Membrane Antigen (EMA), CAM 5.2 and vimentin (Figure 3b). The epithelial markers (AE1/AE3), CK 7, CK 19, and p63 were negative, as well as desmin and melanoma marker S 100. The diagnosis of grade 3 spindle cell monophasic synovial sarcoma with high mitotic index (31 mitosis x10 HPF) was confirmed by the presence of the SS18 (SYT) gene translocation, 18q11, performed by break-apart FISH probe (Figure 3c).
Figure 3: Histopathological examination: a) Hematoxylin-eosin stains showing a monophasic tumor with spindle cells; b) Immunohistochemistry: positivity for vimentin, and c) Break-apart FISH probe: Presence of the SS18 (SYT) gene translocation, 18q11.
Due to the tumor location, a multidisciplinary assessment confirmed the initial unresectability of the lesion. Chemotherapy treatment with ifosfamide 2000mg/m2 and doxorrubicin 20mg/m2 for 3 days every 21 days was initiated with neoadjuvant intention. After three cycles of treatment, surgical option was finally rejected due to disease progression according to RECIST and Choi criteria. High-dose ifosfamide (12g/m2 for 5 days every 21 days) was administrated as a second line treatment, but the patient progress after three doses (Figure 4). A third-line treatment with the tyrosin kinase inhibitor pazopanib 800mg/day was started. After the second cycle, the patient was admitted to the emergency room with progressive dyspnea, orthopnea and hepatalgia. The physical examination revealed bilateral lower limb edema, jugular venous distension, and paradoxical pulse. An echocardiogram confirmed the suspicious cardiac tamponade secondary to constrictive tumoral pericarditis. Seven months after the initial diagnosis of the mediastinal SS, the patient passed away due to this clinical complication. The necropsy confirmed the diagnosis of spindle cell monophasic SS of 25x18x16cm that involved the whole mediastinum and the pulmonary vascular structures and infiltrated the pericardium and the right lung parenchyma. The cause of death was a cardiogenic shock that triggered to a congestive heart failure due to pericardium and right lung infiltration by SS (Figure 5).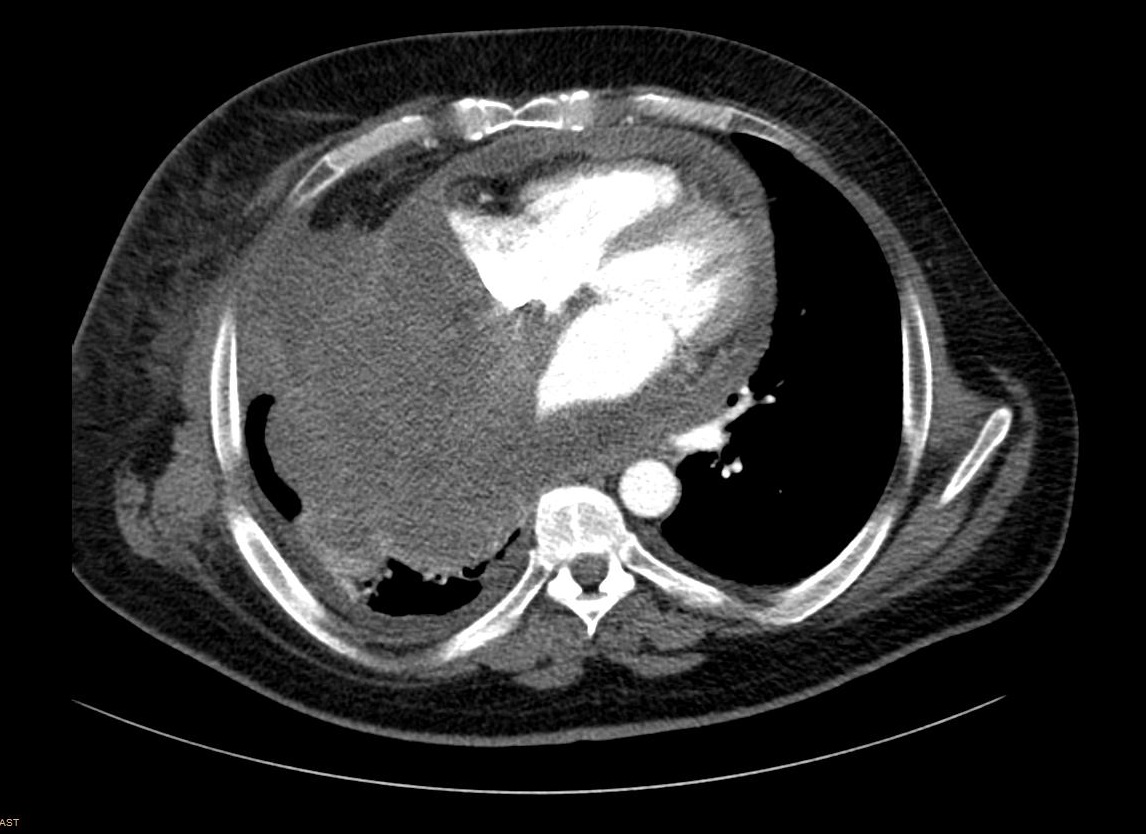
Figure 4: Computed tomography scan showing progression disease with increased right pleural and lung infiltration.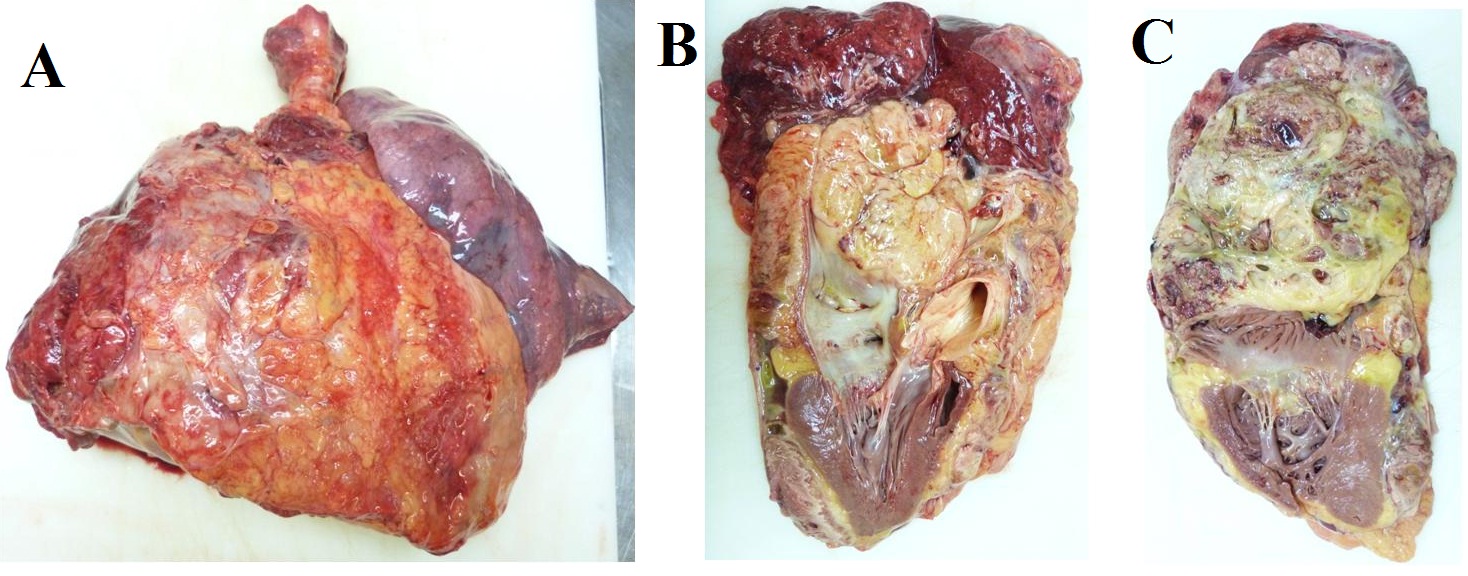
Figure 5: Necropsy. Gross pathology, a) Right lung involved with a huge tumoral mass; b) sagital, and c) axial. Section showing pericardium and atrial infiltration by synovial sarcoma.
DISCUSSION
Our case reflects the high aggressiveness and poor prognosis of mediastinal SS.
With an age at diagnose close to the mean age reported our patient was diagnosed after long time of unspecific symptoms and lived only 7 months, due to the surgery infeasibility.
Despite its name, SS are not derived from synovial tissue. They are named “synovial” for their frequent occurrence in para-articular region, but they can also be found in other locations aloof of joints [18]. SS are characterized by spindle cells with an epithelial origin and they are classified in two different subtypes: monophasic and biphasic or poorly differentiated.
IHC helps to establish the diagnosis. Epithelial markers such as CAM 5.2 and EMA are usually positive. CD99 and S100 are positive in 60% and 40% of the SS, respectively. In our sample S100 was negative. Actin, desmin, CD 34 and CD31 are usually negative in this sarcoma subtype. Up to 90% of cases harbors a SS18 (SYT) gene rearrangement on chromosome 18q11, being this finding a pathognomonic feature for SS [4].
Several studies have evaluated for potential prognostic factors in SS. Despite the fact that the majority of these studies included a heterogeneous population, several factors led to poor prognostic. These include male gender; advanced age at the diagnosis, large tumor size (>5cm), deep location, bone invasion, vascular or perineural invasion, high mitotic activity and overexpression of p53 [5-7].
In our case, size and high mitotic activity appeared to be contributing factors for a poor prognosis, but probably the main negative prognostic factor was the location, that precluded a radical approach.
The diagnosis and treatment of mediastinal sarcoma are challenging. Unlike SS of the limbs, which are usually diagnosed by inspection, mediastinal SS have non-specific symptoms, which usually lead to a delayed diagnosis. When confined, the curative treatment includes surgical removal of the mass with clean margins. The presence of residual disease or affected margins has been related to a bad prognosis. 5 As SS are all considered high grade sarcomas, adjuvant radiotherapy administered as external radiotherapy, brachytherapy or Intensity Modulated Radiotherapy (IMRT) should be applied in tumors over 5cm in size [18].
In cases with unusual location, the proximity to great vessels, heart and lungs prevent curative surgery. Even in selected cases that undergo surgery, the presence of residual disease is common.
As mentioned before, the treatment is often hampered by the tumor location and size. Thus, neoadjuvant chemotherapy could be an option for patients with large tumors considered unresectable at the diagnosis. Although anthracycline-based regimens are still considered the best chemotherapy for STS, SS seem to be especially sensitive to ifosfamide [19-21]. The most used chemotherapy includes high dose ifosfamide or a combination of Adriamycin and ifosfamide. The combination confers response rates of up to 58%, with more chemotherapy-related toxicity. No differences in overall survival have been observed when compared to ifosfamide as monotherapy [22]. Our patient was excluded for surgery due to cardiac infiltration even though he had been treated with a combination of Adriamycin and ifosfamide. Rechallenging with high dose ifosfamide has demonstrated to be active in this sarcoma subtype, even after standard dose [19,20]. Subsequent therapies like gemcitabine alone or in combination, dacarbazine, and new drugs as pazopanib and trabectedin used in unresectable or metastatic setting are quiet efficient in this sarcoma subtype [23-26].
Our case demonstrates the aggressive behavior of a mediastinal SS. An early detection of a SS is crucial to achieve a radical surgery. Other treatments are palliative and the survival of these patients is poor.
REFERENCES
- Zahm SH, Fraumeni JF Jr (1997) The epidemiology of soft tissue sarcoma. Semin Oncol 24: 504-514.
- Cadman NL, Soule EH, Kelly PJ (1965) SYNOVIAL SARCOMA; AN ANALYSIS OF 34 TUMORS. Cancer 18: 613-627.
- Fletcher CDM, Unni KK, Mertens F (2002) Synovial sarcoma. In: Fletcher CDM, Unni KK, Mertens F (eds.). Pathology and Genetics of Tumours of Soft Tissue and Bone, IARC, France. Pg no: 200-204.
- Sandberg AA, Bridge JA (2002) Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors. Synovial sarcoma. Cancer Genet Cytogenet 133: 1-23.
- Lewis JJ, Antonescu CR, Leung DH, Blumberg D, Healey JH, et al. (2000) Synovial sarcoma: a multivariate analysis of prognostic factors in 112 patients with primary localized tumors of the extremity. J Clin Oncol 18: 2087-2094.
- Spillane AJ, A'Hern R, Judson IR, Fisher C, Thomas JM (2000) Synovial sarcoma: a clinicopathologic, staging, and prognostic assessment. J Clin Oncol 18: 3794-3803.
- Trassard M, Le Doussal V, Hacène K, Terrier P, Ranchère D, et al. (2001) Prognostic factors in localized primary synovial sarcoma: a multicenter study of 128 adult patients. J Clin Oncol 19: 525-534.
- Ferrari A, Gronchi A, Casanova M, Meazza C, Gandola L, et al. (2004) Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer 101: 627-634.
- Skinner KA, Eilber FR (1996) Soft tissue sarcoma nodal metastases: biologic significance and therapeutic considerations. Surg Oncol Clin N Am 5: 121-127.
- Salah S, Salem A (2014) Primary synovial sarcomas of the mediastinum: a systematic review and pooled analysis of the published literature. ISRN Oncol 2014: 412527.
- Khan H, Chaubey S, Edlin J, Wendler O (2014) Primary cardiac synovial sarcoma. A rare tumor with poor prognosis. Asian Cardiovasc Thorac Ann 22: 835-838.
- Varma T, Adegboyega P (2012) Primary cardiac synovial sarcoma. Arch Pathol Lab Med 136: 454-458.
- Phatak P, Khanagavi J, Aronow WS, Puri S, Yusuf Y, et al. (1000) Pericardial synovial sarcoma: challenges in diagnosis and management. Version 2, F1000Res 3: 15.
- Salah S, Al-Ibraheem A, Daboor A, Al-Hussaini M (2013) Synovial sarcoma presenting with huge mediastinal mass: a case report and review of literature. BMC Res Notes 6: 240.
- Boulmay B, Cooper G, Reith JD, Marsh R (2007) Primary cardiac synovial sarcoma: a case report and brief review of the literature. Sarcoma 2007: 94797.
- Zhang L, Qian J, Li Z, Jing H (2011) Primary synovial sarcoma of the heart. Cardiol J 18: 128-133.
- Gaetano Reaa, Francesco Sommab, Tullio Valentea, Giuseppe Antinolfic, Graziella Di Grezia, et al. (2015) Primary mediastinal giant synovial sarcoma: A rare case report. The Egyptian Society of Radiology and Nuclear Medicine 46: 9-12.
- Eilber FC, Dry SM (2008) Diagnosis and management of synovial sarcoma. J Surg Oncol 97: 314-320.
- ESMO/European Sarcoma Network Working Group (2014) Soft tissue and visceral sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 3: 102-112.
- Le Cesne A, Antoine E, Spielmann M, Le Chevalier T, Brain E, et al. (1995) High-dose ifosfamide: circumvention of resistance to standard-dose ifosfamide in advanced soft tissue sarcomas. J Clin Oncol 13: 1600-1608.
- Rosen G, Forscher C, Lowenbraun S, Eilber F, Eckardt J, et al. (1994) Synovial sarcoma. Uniform response of metastases to high dose ifosfamide. Cancer 73: 2506-2511.
- Judson I, Verweij J, Gelderblom H, Hartmann JT, Schöffski P, et al. (2014) Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: a randomised controlled phase 3 trial. Lancet Oncol 15: 415-423.
- Maki RG, Wathen JK, Patel SR, Priebat DA, Okuno SH, et al. (2007) Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 25: 2755-2763.
- García-Del-Muro X, López-Pousa A, Maurel J, Martín J, Martínez-Trufero J, et al. (2011) Randomized phase II study comparing gemcitabine plus dacarbazine versus dacarbazine alone in patients with previously treated soft tissue sarcoma: a Spanish Group for Research on Sarcomas study. J Clin Oncol 29: 2528-2533.
- van der Graaf WT, Blay JY, Chawla SP, Kim DW, Bui-Nguyen B, et al. (2012) Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial below Lancet 379: 1879-1886.
- Demetri GD, Chawla SP, von Mehren M, Ritch P, Baker LH, et al. (2009) Efficacy and safety of trabectedin in patients with advanced or metastatic liposarcoma or leiomyosarcoma after failure of prior anthracyclines and ifosfamide: results of a randomized phase II study of two different schedules. J Clin Oncol 27: 4188-4196.
Citation: Estival A, Balaña C, Hardy M (2015) A 38 Year-old Man with Mediastinal Synovial Sarcoma: Case Report and Review of Literature. J Clin Stud Med Case Rep 2: 021.
Copyright: © 2015 Anna Estival, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.