Journal of Nephrology & Renal Therapy Category: Clinical
Type: Case Report
A Case Report of a Novel Variant of X-linked Alport Syndrome
*Corresponding Author(s):
Robert M GaetaNephrology Service, Department Of Medicine, Walter Reed National Military Medical Center, Bethesda, Maryland, United States
Tel:+1 3012954331,
Email:robert.m.gaeta.mil@mail.mil
Received Date: Jun 14, 2018
Accepted Date: Jul 20, 2018
Published Date: Aug 09, 2018
Abstract
X-linked Alport syndrome is a rare hereditary disorder caused by variants of the COL4A5 gene. We describe a case of a 28 year old Caucasian male with a family history of end-stage renal disease presenting with episodic gross hematuria and nephrotic range proteinuria. Renal biopsy demonstrated focal segmental glomerulosclerosis with non-diagnostic ultrastructural findings. Next Generation Sequencing revealed a COL4A5 missense likely pathogenic variant, a substitution of adenine for guanine at nucleotide 901(c.901G>A) of the coding DNA predicting a glycine to serine substitution at amino acid 301 (p.Glyc301Ser). This variant has not been reported in literature or human genomic databases.
Keywords
Alport; FSGS; Missense; Variant, X-linked
BACKGROUND
Alport Syndrome (AS) is a rare hereditary disorder caused by variants of the COL4A3-5 genes, with 85% X-linked (Xq22) on COL4A5 [1]. The syndrome is characterized by progressive nephritis with ultrastructural changes of the glomerular basement membrane, frequently associated sensorineural hearing loss, and occasional ophthalmologic disease, lenticonus or retinal flecks [1]. Renal histology presents on a spectrum from archetypal ultrastructural lamellated basement membranes (~62%) to minimal ultrastructural findings (~12%) often with Focal Segmental Glomerulosclerosis (FSGS) on light microscopy [2,3] (Figure 1). The latter category represented a diagnostic challenge prior to the clinical application of Next Generation Sequencing (NGS). Remarkably, COL4A3-5 variants represent a common etiology of familial FSGS (38%) with over half of these mutations in COL4A5 [3]. Here we describe a case of familial FSGS diagnosed with a novel COL4A5 variant.
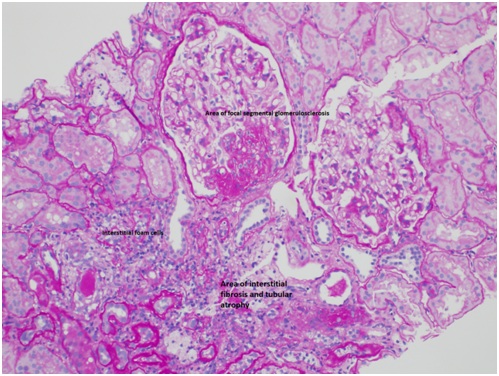
Figure 1: Alport missense mutation.
CASE REPORT
A 28 year-old caucasian male with a family history of End-Stage Renal Disease (ESRD) presented with episodic gross hematuria and nephrotic range proteinuria. He reported four lifetime episodes of gross hematuria beginning five years ago. He also described frothy urine and one remote episode of lower extremity swelling. He developed sustained hypertension six months prior to presentation. He had been regularly prescribed NSAIDs for the last eight years. He denied any supplement, anabolic steroid, or illicit drug use. He had no history of hearing loss, confirmed on formal serial annual evaluations, and a recent unremarkable ophthalmology exam. His only other medications were prescribed for seasonal allergies.
Past medical history was notable for compartment syndrome requiring bilateral lower extremity compartment release six years ago. He is an active duty service member and completed two prior deployments. His mother progressed to ESRD due to an unknown etiology at age 35, which prompted a formal pedigree evaluation (Figure 2). His maternal grandfather died at 50, and three maternal great uncles had known kidney disease. No one in the family was previously biopsied. Blood pressure was 146/92 mmHg. Physical examination was unremarkable with no lower extremity edema.
Past medical history was notable for compartment syndrome requiring bilateral lower extremity compartment release six years ago. He is an active duty service member and completed two prior deployments. His mother progressed to ESRD due to an unknown etiology at age 35, which prompted a formal pedigree evaluation (Figure 2). His maternal grandfather died at 50, and three maternal great uncles had known kidney disease. No one in the family was previously biopsied. Blood pressure was 146/92 mmHg. Physical examination was unremarkable with no lower extremity edema.
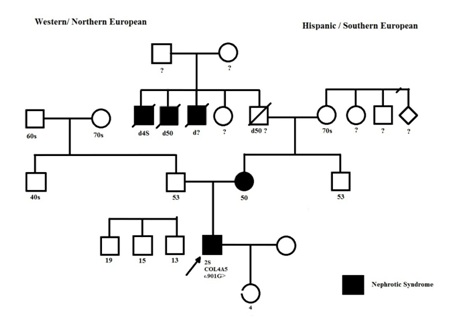
Figure 2: Pedigree structure. Proband is arrowed. Squares represent males, circles represent females. Family history is relevant for ESRD and nephritic syndrome in his mother and three maternal great-uncles. His Maternal grandfather had a history of kidney disease, without ESRD, and passed away at the age of 50 years from an unknown etiology. The proband has a 4-year-old daughter his history of febrile seizures, allergies and asthma. Her carrier status for COL4A5 c.901G>A (p.Gly301Ser) is unknown. She has no history of hematuria.
His first Urinalysis (UA) five years prior to presentation and all subsequent UAs demonstrated 3+ blood and 2-3+ proteinuria. Urine sediment analysis on presentation was remarkable for 60-100 dysmorphic RBCs. His serum creatinine ranged from 1-1.3 mg/dl for all available data points over the last 5 years, with cystatin-C level of 0.63 mg/L on presentation (CKD-EPI Creatinine-Cystatin C eGFR 102ml/min/1.73m2). Serum albumin was 4.1g/dL and he had 7.7gm/24 hour of urine protein. Serum complement was negative and HIV screening negative. A renal ultrasound was unremarkable.
Because of nephrotic-range proteinuria and strong family history of renal disease, renal biopsy and genetic testing were performed. Renal biopsy demonstrated FSGS with non-diagnostic ultrastructural findings (Figure 3). NGS and Sanger Sequencing revealed a COL4A5 missense variant, a substitution of adenine for guanine at nucleotide 901(c.901G>A) of the coding DNA predicting a glycine to serine substitution at amino acid 301 (p.Glyc301Ser). The clinical genetic testing was performed at Prevention Genetics, LLC (Marshfield, WI). This variant has not been reported in the literature or in databases cataloguing natural human genetic variation including dbSNP and the 1000 Genomes project.

Figure 3: Electron Microscopy: Shows foot process effacement, mild rarefaction and thick basement membranes (measurements included). Light microscopy (not pictured) demonstrated 3 of 13 glomeruli with segmental scarring and 15-20% interstitial fibrosis and tubular atrophy. Immunofluorescence (not pictured) was only positive for non-specific arteriolar and mesangial C3.
DISCUSSION
The pathogenicity for the missense variant identified is strongly suspected based on its location in a conserved GLY-Xaa-Yaa triple helical domain in the COL4A5 gene. Knebelmann et al., first reported a similar missense variant substituting arginine for glycine at position 325 in large kindred of X-linked Alport syndrome [4]. The GLY-Xaa-Yaa repeat is characteristic of the collagenous domain, and is normally interrupted 22 times by non-collagenous sequences [4]. A missense variant would be expected to add an additional interruption of the triple helix conformation, which relies on the presence of glycine, the smallest amino acid, to form the center of the helix [4]. In our case, the serine for glycine substitution creates an additional collagen domain interruption at position 301 in the alpha 5 chain of type IV collagen.
The variant genotype has significant implications for individual prognosis. A study of 175 X-linked AS families demonstrated that rate of progression of renal and extra-renal manifestations was associated with the type of mutation, where those with Gly-Xaa-Yaa variants reached ESRD at a median of 33 years compared with 25 years for truncating mutations [5]. In contrast to non-Gly-Xaa-Yaa variants, there was no relationship between mutation position and age of ESRD in those with Gly-Xaa-Yaa variants [5]. This variant also has implications for COL4A3-5 expression (~25% of AS patients) [2] and we expect expression marks a lower risk for post renal transplant anti-glomerular basement membrane disease.
In conclusion, genetic testing is essential for appropriate management of familial FSGS. We report a case of X-linked AS due to a novel COL4A5 variant detected by NGS. Correct diagnosis allowed for appropriate genetic counseling and treatment, including ACE inhibitor therapy and the avoidance of immunosuppression. Diagnosis also facilitated appropriate disposition to a medical evaluation board given his expected progression to ESRD and high risk for later hearing loss [6].
The variant genotype has significant implications for individual prognosis. A study of 175 X-linked AS families demonstrated that rate of progression of renal and extra-renal manifestations was associated with the type of mutation, where those with Gly-Xaa-Yaa variants reached ESRD at a median of 33 years compared with 25 years for truncating mutations [5]. In contrast to non-Gly-Xaa-Yaa variants, there was no relationship between mutation position and age of ESRD in those with Gly-Xaa-Yaa variants [5]. This variant also has implications for COL4A3-5 expression (~25% of AS patients) [2] and we expect expression marks a lower risk for post renal transplant anti-glomerular basement membrane disease.
In conclusion, genetic testing is essential for appropriate management of familial FSGS. We report a case of X-linked AS due to a novel COL4A5 variant detected by NGS. Correct diagnosis allowed for appropriate genetic counseling and treatment, including ACE inhibitor therapy and the avoidance of immunosuppression. Diagnosis also facilitated appropriate disposition to a medical evaluation board given his expected progression to ESRD and high risk for later hearing loss [6].
ACKNOWLEDGEMENT
Informed consent was obtained. The clinical genetic testing was performed at Prevention Genetics, LLC (Marshfield, WI). A complete pedigree assessment was performed by Ms. Meagan T. Monte MS CGC, genetic counselor. The views expressed in this report are those of the authors, and do not reflect the official policy of the Department of the Army, the Department of the Navy, the Department of Defense, or the United States Government.
REFERENCES
- Savige J, Gregory M, Gross O, Kashtan C, Ding J, et al. (2013) Expert Guidelines for the Management of Alport Syndrome and Thin Basement Membrane Nephropathy. J Am Soc Nephrol 24: 364-375
- Mazzucco G, Barsotti P, Muda AO, Fortunato M, Mihatsch M, et al. (1998) Ultrastructural and immunohistochemical findings in Alport's syndrome: a study of 108 patients from 97 Italian families with particular emphasis on COL4A5 gene mutation correlations. J Am Soc Nephrol 9: 1023-1031.
- Gast C, Pengelly RJ, Lyon M, Bunyan DJ, Seaby EG, et al. (2016) Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol Dial Transplant 31: 961-970.
- Knebelmann B, Deschenes G, Gros F, Hors MC, Grünfeld JP, et al. (1992) Substitution of arginine for glycine 325 in the collagen alpha 5 (IV) chain associated with X-linked Alport syndrome: characterization of the mutation by direct sequencing of PCR-amplified lymphoblast cDNA fragments. Am J Hum Genet 51: 135-142.
- Bekheirnia MR, Reed B, Gregory MC, McFann K, Shamshirsaz AA, et al. (2010) Genotype-phenotype correlation in X-linked Alport syndrome. J Am Soc Nephrol 21: 876-883.
- Jais JP, Knebelmann B, Giatras I, De Marchi M, Rizzoni G, et al. (2000) X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J Am Soc Nephrol 11: 649-657.
Citation: Gaeta RM, Childs M, Datta AA, Veras JE, Nee R, et al. (2018) A Case Report of a Novel Variant of X-linked Alport Syndrome. J Nephrol Renal Ther 4: 017.
Copyright: © 2018 Robert M Gaeta, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Journal Highlights
© 2026, Copyrights Herald Scholarly Open Access. All Rights Reserved!