A Lynch-Like Presentation of Birt-Hogg-Dube Syndrome (BHDS)
*Corresponding Author(s):
Rachael BrandtMain Line Health, Lankenau Institute For Medical Research, Thomas Jefferson University, Pennsylvania, United States
Tel:+1 4844768153,
Email:BrandtR@MLHS.ORG
Abstract
Introduction
Over the past few years, next generation sequencing technology and the use of panel genetic testing has quickly replaced traditional single gene testing using Sanger sequencing. While gene panels are capable of testing for multiple genetic conditions simultaneously thereby decreasing overall costs, personal and family history still play an important role in determining an appropriate gene panel and without attention in laboratory and test selection, rare syndromes may be missed.
Case report
Multiple members from a family affected with Lynch-syndrome cancers and meeting Amsterdam II criteria presented nearly simultaneously for genetic testing based on personal history, family history and absent immunohistochemical staining in select tumors. Careful review of the pathology and of the family history, including benign findings, altered the initial test approach for the family. A pathogenic FLCN mutation, consistent with Birt-Hogg-Dube syndrome (BHDS) was identified in multiple family members.
Discussion
This case illustrates the utility of panel testing in comprehensive genetic evaluations. Due to the pre-test clinical suspicion of multiple cancer predisposition syndromes, this family was an excellent example of an ideal clinical scenario for panel testing. By submitting an expanded panel test for these family members, differential diagnoses were assessed simultaneously, and overall cost was lower. Findings significantly impacted the medical management guidelines for individuals identified to have BHDS.
INTRODUCTION
Over the past several decades, genetic testing for hereditary cancer predisposition syndromes has become standard of care in oncology management. The American Society of Clinical Oncology has endorsed inherited susceptibility testing as a part of cancer management since 1996 [1]. The primary goals of testing include enabling patients to make choices on cancer screening, surgical and chemopreventive risk-reduction and targeted treATMent options, which has been associated with a lower incidence of both cancer and overall mortality among those testing gene-positive [2].
Traditionally, genetic testing involved testing a single gene at a time, primarily using Sanger sequencing [3]. More recently, the field has adopted next generation sequencing technology which has led to multigene panel testing needs replacing single gene tests and multi-gene panel genetic testing has nearly replaced single gene testing. Gene panels are capable of testing for multiple genetic conditions simultaneously, thereby decreasing overall costs and obtaining results faster than the single gene method [4,5]. Additional advantages of panel testing include increased sensitivity for cancer predisposition syndromes. Numerous studies have established that panel testing for breast, colorectal and/or ovarian cancer indications identifies more patients with gene mutations in less common or less penetrant genetic syndromes that may not have been identified based on clinical and/or insurance testing criteria [6,7]. This can be especially useful for testing in cases that lack distinguishing clinical characteristics. Thus, the National Comprehensive Cancer Network (NCCN) currently endorses the use of multi-gene panel testing as an appropriate test for certain patients in the context of pre- and post-test guidance from a genetics professional [8].
With the use of cross-cancer panels, analysis of genes beyond those clinically indicated has become increasingly common and can yield unexpected findings. For example, a woman presenting for genetic evaluation due to personal history of triple negative breast cancer was found to carry a mutation in the gene MLH1, consistent with a diagnosis of Lynch syndrome [9]. Further, an analysis of 1112 patients clinically appropriate for Lynch syndrome testing revealed a BRCA1/2 mutation in 15 patients [10]. Though unexpected, such results serve to promote further characterization of known genetic syndromes and the personalization of appropriate risk-based management guidelines beyond that which could be established from pedigree analysis alone had the traditional single gene test approach been employed.
In this report, panel testing proved to be essential in the successful management of family X. This family was identified for evaluation through a community-based hospital system’s universal Immunohistochemical (IHC) staining program of colorectal and endometrial cancer specimens, designed to identify patients at high-risk for Lynch syndrome. Per program protocol, patients with absent staining by IHC are offered further evaluation by a genetic counselor that includes review of pedigree and discussion regarding genetic test options.
CASE REPORT
The first member of family X to present to the program was a 91 year old male, individual II.4, with a poorly differentiated adenocarcinoma of the ascending colon with metastatic disease to the liver (Figure 1). IHC showed loss of MLH1 and PMS2. Additional personal cancer history included three metachronous bladder cancers between his 70s and 90 and a history of melanoma at age 86. He was a prior smoker (pack year history unknown). Further tumor analysis for BRAF V600E and/or hypermethylation or germline testing was unable to be completed prior to his unexpected death secondary to complications.
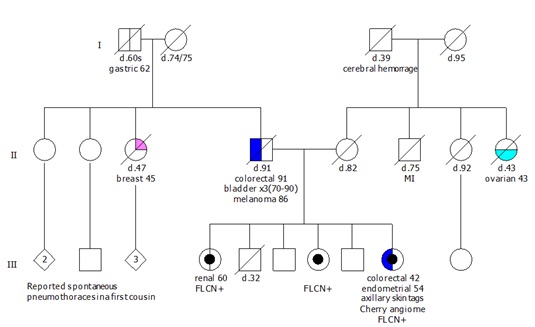 Figure 1: Pedigree.
Figure 1: Pedigree.
Several months later, individual II.4’s daughter, individual III.9 presented with a diagnosis of endometrioid adenocarcinoma at age 54. Immunohistochemical staining of the mismatch repair proteins was performed and showed loss of MSH6 protein staining. The MLH1, MSH2 and PMS2 protein staining was intact. Additional history included personal history of a right sided colon cancer with liver metastases at age 41 treated with an open right hemicolectomy with partial left hepatic resection for metastasis and adjuvant FOLFOX chemotherapy, currently without evidence of recurrence, a family history of renal cancer in her sister and of gastric cancer in her paternal grandfather.
Individual III.9 declined further genetic evaluation, however, her sister with renal cancer, individual III.4, elected to pursue this. She reported to the program a personal history of two tumors of the right kidney at age 59. Pathology was obtained and confirmed a chromophobe renal cell carcinoma in the middle POLE(2.5cm) and a multilocular cystic tumor, in the lower POLE combined chromophobe and cystic clear cell carcinoma. An adrenal cortical adenoma was also noted on surgical specimen. Given the renal pathology and family history, a genetic panel was ordered that included MMR genes, FLCN and other genes associated with renal and/or colorectal/endometrial cancer. A pathogenic FLCN mutation, c.557G>A (p.Trp186*), and a variant of uncertain significance, c.3857G>A in POLE were identified. This mutation in FLCN truncates the protein prematurely at codon 186 and is classified as pathogenic in the ClinVar database (Variation ID: 230137). No reported mutations/variants were identified in any of the other genes analyzed by sequencing and/or deletion/duplication analysis: APC, ATM, AXIN2, BAP1, BARD1, BMPR1A, BRCA1, BRCA2, BRIP1, CDH1, CDK4, CDKN2A, CHEK2, EPCAM,FANCC, FH, GREM1,MEN1, MET, MLH1, MRE11A, MSH2, MSH6, MUTYH, NBN, NF1, PALB2, PMS2, POLD1, PTEN, RAD50, RAD51C, RAD51D, SDHA, SDHB, SDHC, SDHD, SMAD4, SMARCA4, STK11, TP53, TSC1, TSC2, VHL or XRCC2. Subsequent evaluation of individual III.4’s medical records revealed acrochordons and a history of lung cysts. A history of fibrofolliculomas or colorectal polyps was denied.
Upon identification of the FLCN mutation in the family, individual III.9 reconsidered genetic evaluation. Individual III.9 reported a history of bilateral axillary acrochordons (skin tags). A report from a CT scan of the chest, abdomen and pelvis from 4 years prior showed a stable pattern of scattered bilateral pulmonary bullae and blebs (cysts) and a stable subcentimeter left renal cortical hypodensity. A 43 gene hereditary cancer panel was ordered, which confirmed presence of the FLCN c.557G>A mutation. She tested negative for the POLE VUS and no mutations were identified in any of the Lynch syndrome genes, including MSH6, or other genes tested.
Additionally, individual III.4’s other sister, individual III.7, presented for testing. She reported a personal history of several skin lesions (nasal angiofibroma/fibrous papule, basal cell carcinoma of the left distal pretibia and intradermal nevus of the neck, verrucous keratosis and basal cell carcinoma of the left arm) and 3 colorectal polyps, type unknown. Multi-gene panel testing was performed and the FLCN mutation was also identified (Table 1).
|
Individual |
Clinical Finding |
Genetic Test Result |
|
II.4 |
Colorectal cancer |
Unknown |
|
Melanoma |
||
|
Bladder x3 |
||
|
III.4 |
Chromophobe renal cell cancer |
FLCN+ |
|
Acrochordons |
||
|
Lung cysts |
||
|
III.7 |
Several skin lesions (nasal angiofibroma/fibrous papule, basal cell carcinoma and intradermal nevus of the neck, verrucous keratosis and basal cell carcinoma) |
FLCN+ |
|
3 colorectal polyps, type unknown |
||
|
III.9 |
Endometrial cancer |
FLCN+ |
|
Colorectal cancer |
||
|
Bilateral axillary acrochordons scattered bilateral pulmonary bullae and blebs (cysts) |
||
|
Stable subcentimeter left renal cortical hypodensity |
Table 1: Clinical manifestations reported in family X.
Given family X’s presentation, additional testing of colorectal tumor DNA is under consideration in an effort to provide explanation of the absent IHC staining observed in family members affected with colorectal cancer. While lineage of the FLCN mutation has not yet been established, a paternal first cousin has been reported to have had multiple spontaneous pneumothoraces.
DISCUSSION
Birt-Hogg-Dubé Syndrome (BHDS) is a rare cancer predisposition syndrome first described by three Canadian physicians in 1977 [11]. BHDS is inherited in an autosomal dominant manner and is caused by loss-of-function pathogenic mutations in the gene Folliculin (FLCN) [12]. Approximately 91-93% of individuals meeting clinical diagnostic criteria for BHDS will have an identifiable mutation in FLCN by sequencing or deletion/duplication analysis [13].
BHDS is primarily characterized by three features: benign cutaneous manifestations, pulmonary cysts and spontaneous pneumothorax and renal tumors. Affected individuals vary significantly in presentation and severity, even within families. Skin growths include fibrofolliculomas, trichodiscomas/angiofibromas, perifollicular fibromas and acrochordons [14,15]. These typically appear when individuals are in their twenties or thirties and can increase in size and number over time. Lung cysts are mostly bilateral and multifocal and have been observed in up to 89% [16] of individuals with pathogenic FLCN mutations; most individuals are asymptomatic but have a high risk for spontaneous pneumothorax [17]. Reports for renal involvement vary, as 12% to 34% of individuals with BHDS develop renal (kidney) tumors [17,18], which also tend to be bilateral and multifocal and are usually slow growing; median age of renal tumor diagnosis is 48 years.
Additional findings of BHDS continue to be characterized. Since 2000, several affected individuals have been reported with parotid oncocytomas [19,20]. Other features such as thyroid cancer or nodules and oral papules have also been reported [14,21]. Early reports of BHDS included intestinal polyps and colorectal cancer as an associated feature of the condition [17,22].
Further studies concerning the BHDS clinical spectrum have failed to consistently establish colonic neoplasms as a clinical feature [23,24]. Recent work has suggested the possibility of genotype/phenotype correlations underlying the risk for colorectal findings in BHDS [25] with carriers of mutation FLCNc.1285dupC displaying a significantly higher risk for colorectal neoplasia compared to carriers of FLCNc.610delGCinsTA. Toro et al., [20] additionally reported occurrences of Squamous Cell Carcinoma (SCC) of the head, neck, and cervix, Hodgkin’s disease, uterine cancer, prostate cancer, breast cancer, rhabdomyoma and an adrenal mass in their study of 51 families with BHDS; none of these findings, however, have been confirmed to be part of the BHDS clinical spectrum to date.
SUMMARY POINTS
Family X’s initial presentation, of gastric, colorectal and endometrial cancers met Amsterdam II criteria [26] for possible Lynch syndrome. This suspicion was furthered by the loss of MSH6 in III.9 endometrial cancer specimen. It was not until III.4 presented and renal pathology reports were obtained that the possibility of a familial FLCN mutation was raised. This serves to highlight the necessity of accurate and complete family histories when pursing genetic testing. Without this information, this family would not have met criteria for FLCN gene testing, and may not have been evaluated for this rare syndrome. Due to the pre-test clinical suspicion of multiple cancer predisposition syndromes, family X was an excellent example of an ideal clinical scenario for panel testing. By submitting panel testing for these patients, the main concerns on the differential diagnosis were assessed simultaneously, and overall cost was lower. Additionally, as the clinical associations of cancer syndromes remains under investigation, panel testing allows for further elucidation of germline mutation risks in families that may not have a traditional presentation. This type of result and case report is an opportunity to revisit the known phenotype of FLCN mutation carriers.
Had individual III.4 not presented for genetic evaluation and permitted release of her renal cancer pathology, the chromophobe renal cancers would not have been confirmed. This pathology directed the testing plan for the family; testing of FLCN would not have been considered based on III.9 personal or reported history at that time. Furthermore, she would not have met clinical testing criteria for Birt-Hogg-Dubé syndrome and depending on the lab used or panel ordered, FLCN may not have been included. In fact, many healthcare providers may have ordered only MSH6 testing given the abnormal IHC result in the endometrial tissue and not considered any further genes for evaluation. Not only can panel testing reveal unexpected genetic test results, it can be strategically used for a more thorough genetic evaluation or to test genes that may not have been able to be tested independently.
REFERENCES
- Robson ME, Bradbury AR, Arun B, Domchek SM, Ford JM, et al. (2015) American society of clinical oncology policy statement update: genetic and genomic testing for cancer susceptibility. J Clin Oncol 33: 3660-3667.
- Domchek SM, Friebel TM, Singer CF, Evans DG, Lynch HT, et al. (2010) Association of risk-reducing surgery in BRCA1 or BRCA2 mutation carriers with cancer risk and mortality. JAMA 304: 967-975.
- Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 74: 5463-5467.
- Pritchard CC, Smith C, Salipante SJ, Lee MK, Thornton AM (2012) ColoSeq provides comprehensive lynch and polyposis syndrome mutational analysis using massively parallel sequencing. J Mol Diagn 14: 357-366.
- Walsh T, Lee MK, Casadei S, Thornton AM, Stray SM, et al. (2010) Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci USA 107: 12629-12633.
- LaDuca H, Stuenkel AJ, Dolinsky JS, Keiles S, Tandy S, et al. (2014). Utilization of multigene panels in hereditary cancer predisposition testing: analysis of more than 2,000 patients. Genet Med 16: 830-837.
- Maxwell KN, Wubbenhorst B, D’Andrea K, Garman B, Long JM, et al. (2015) Prevalence of mutations in a panel of breast cancer susceptibility genes in BRCA1/2-negative patients with early-onset breast cancer. Genet Med 17: 630-638.
- Daly MB, Pilarski P, Berry M, Buys SS, Farmer M, et al. (2017) NCCN Clinical practice guidelines in oncology: Genetic / Familial high-risk assessment: breast and ovarian version 2.2017. J Natl Compr Canc Netw 15: 9-20.
- Kurian AW, Hare EE, Mills MA, Kingham KE, McPherson L, et al. (2014) Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J Clin Oncol 32: 2001-2009.
- Yurgelun MB, Allen B, Kaldate RR, Bowles KR, Judkins T, et al. (2015) Identification of a variety of mutations in cancer predisposition genes in patients with suspected lynch syndrome. Gastroenterology 149: 604-613.
- Birt AR, Hogg GR, Dubé WJ (1977) Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol 113: 1674-1677.
- Nickerson ML, Warren MB, Toro JR, Matrosova V, Glenn G, et al. (2002) Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dubé Cancer Cell 2: 157-164.
- Schmidt LS, Lineham WM (2015) Molecular genetics and clinical features of Birt-Hogg-Dubé Nature Reviews Urology 12: 558-569.
- Kunogi M, Kurihara M, Ikegami TS, Kobayashi T, Shindo N, et al. (2010) Clinical and genetic spectrum of Birt-Hogg-Dube syndrome patients in whom pneumothorax and/or multiple lung cysts are the presenting feature. J Med Genet 47: 281-287.
- Vernooij M, Claessens T, Luijten M, van Steensel MA, Coull BJ (2013) Birt-Hogg-Dubé syndrome and the skin. Fam Cancer 12: 381-385.
- Toro JR, Pautler SE, Stewart L, Glenn GM, Weinreich M, et al. (2007) Lung cysts, spontaneous pneumothorax, and genetic associations in 89 families with Birt-Hogg-Dubé Am J Respir Crit Care Med 175: 1044-1053.
- Rongioletti F, Hazini R, Gianotti G, Rebora A (1989) Fibrofolliculomas, tricodiscomas and acrochordons (Birt-Hogg-Dubé) associated with intestinal polyposis. Clin Exp Dermatol 14: 72-74.
- Houweling AC, Gijezen LM, Jonker MA, van Doorn MBA, Oldenburg RA, et al. (2011) Renal cancer and pneumothorax risk in Birt–Hogg–Dubé syndrome; an analysis of 115 FLCN mutation carriers from 35 BHD families. Br J Cancer 105: 1912-1919.
- Liu V, Kwan T, Page EH (2000) Parotid oncocytoma in the Birt-Hogg-Dubé J Am Acad Dermatol 43: 1120-1122.
- Toro JR1, Wei MH, Glenn GM, Weinreich M, Toure O (2008) BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet 45: 321-31.
- Kluger N, Giraud S, Coupier I, Avril MF, Dereure O, et al. (2010) Birt- Birt-Hogg-Dubé syndrome: clinical and genetic studies of 10 French families. Br J Dermatol 162: 527-537.
- Hornstein OP, Knickenberg M (1975) Perifollicular fibromatosis cutis with polyps of the colon--a cutaneo-intestinal syndrome sui generis. Arch Dermatol Res 253: 161-175.
- Toro JR1, Glenn G, Duray P, Darling T, Weirich G (1999) Birt-Hogg-Dubé syndrome: a novel marker of kidney neoplasia. Arch Dermatol 135: 1195-1202.
- Zbar B, Alvord WG, Glenn G, Turner M, Pavlovich CP (2002) Risk of renal and colonic neoplasms and spontaneous pneumothorax in the Birt-Hogg-Dubé Cancer Epidemiol Biomarkers & Prev 11: 393-400.
- Nahorski MS, Lim DH, Martin L, Gille JJ, McKay K, et al. (2010) Investigation of the Birt-Hogg-Dube tumour suppressor gene (FLCN) in familial and sporadic colorectal cancer. J Med Genet 47: 385-390.
- Vasen HF, Watson P, Mecklin JP, Lynch HT (1999) New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology 116: 1453-1456.
Citation: Rogers C, Klein A, Stumm K, Nixon C, Brandt R, et al. (2017) A Lynch-Like Presentation of Birt-Hogg-Dube Syndrome (BHDS). J Community Med Public Health Care 4: 032.
Copyright: © 2017 Carolyn Rogers, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.