Journal of Gastroenterology & Hepatology Research Category: Medical
Type: Case Report
A Novel Manifestation of Prolidase Deficiency in a Toddler Diagnosed with Very-Early Onset Crohn’s Disease
*Corresponding Author(s):
S. Ahsan RizviDepartment Of Pediatrics, UF Health Shands Children’s Hospital, University Of Florida, 1600 SW Archer Rd , Gainesville, FL 32608, United States
Tel:352-273-9001,
Fax:352-294-5247
Email:Ahsanraza1989@gmail.com, elderme@peds.ufl.edu, genie@peds.ufl.edu
Received Date: Jun 28, 2019
Accepted Date: Aug 06, 2019
Published Date: Aug 13, 2019
Abstract
Prolidase deficiency is a rare autosomal recessive disorder that presents with recurrent skin ulcers with facial dimorphism and mental retardation. Though, coexistence of prolidase deficiency with Systemic Lupus Erythematous has been well described in literature, the disease is usually not associated with inflammatory bowel disease. We present a novel case of a patient who presented with scrotal swelling and was discovered to have Crohn’s disease with co-existence of Prolidase deficiency. When considering for differential diagnosis of recurrent skin ulcers in setting of IBD, Prolidase deficiency should be considered.
Keywords
Metastatic crohn’s disease; Prolidase deficiency; Chronic diarrhea; Whole exome sequencing; Immunology
ABBREVIATIONS
VEO-IBD: Very Early Onset Inflammatory Bowel Disease
PD: Prolidase Deficiency
MCD: Metastatic Crohn’s Disease
PD: Prolidase Deficiency
INTRODUCTION
Prolidase deficiency (PD) is an autosomal recessive disorder of proline and hydroxproline metabolism. It is usually associated with cutaneous ulcers, dysmorphic facies, skeleton deformities, hematological anomalies, splenomegaly and recurrent chronic infections. The first case of PD was described by Goodman et al, in 1968 in a male patient who had non-healing recurrent ulcers on the lower legs and mental retardation[1]. PD is an extremely rare disorder with overall incidence of 1-2 per 1000,000 individuals; so far only 93 have been reported in literature[2].
Due to the rarity of this disease, there is insufficient knowledge available and therefore further investigation is needed as it may possibly be associated with inflammatory diseases such as Crohn’s disease. We report a case that illustrated extremely rare association of Prolidase deficiency and very-early onset Crohn’s disease in the same patient; the rare combination has only been described in literature once[3-4].
Crohn’s disease is a chronic inflammatory, granulomatous bowel disorder that can affect the gastrointestinal tract from mouth to anus[5]. Diagnosis of Crohn’s disease can be extremely challenging in a pediatric patient as the clinical manifestations are highly variable[6]. Although the average age of diagnosis of Pediatric IBD is 10 years to 12 years, the fastest rates of rise in new diagnosis have been in younger age groups, including those <4 years[6-8]. Very-early-onset IBD is a subset of inflammatory bowel disease accounts of 15% of pediatric IBD cases who present with symptoms and diagnosis prior to 5 years of age. VEO-IBD generally have a worse prognosis as they may not respond to conventional therapies and can include primary immunodeficiencies with GI manifestation. Whole-exome sequencing has been crucial in identifying these novel variants[9].Our patient’s diagnosis of Crohn’s disease was delayed mostly due to his young age, the complexity of diagnosis in his age group and the co-morbidities associated with his disease.
Due to the rarity of this disease, there is insufficient knowledge available and therefore further investigation is needed as it may possibly be associated with inflammatory diseases such as Crohn’s disease. We report a case that illustrated extremely rare association of Prolidase deficiency and very-early onset Crohn’s disease in the same patient; the rare combination has only been described in literature once[3-4].
Crohn’s disease is a chronic inflammatory, granulomatous bowel disorder that can affect the gastrointestinal tract from mouth to anus[5]. Diagnosis of Crohn’s disease can be extremely challenging in a pediatric patient as the clinical manifestations are highly variable[6]. Although the average age of diagnosis of Pediatric IBD is 10 years to 12 years, the fastest rates of rise in new diagnosis have been in younger age groups, including those <4 years[6-8]. Very-early-onset IBD is a subset of inflammatory bowel disease accounts of 15% of pediatric IBD cases who present with symptoms and diagnosis prior to 5 years of age. VEO-IBD generally have a worse prognosis as they may not respond to conventional therapies and can include primary immunodeficiencies with GI manifestation. Whole-exome sequencing has been crucial in identifying these novel variants[9].Our patient’s diagnosis of Crohn’s disease was delayed mostly due to his young age, the complexity of diagnosis in his age group and the co-morbidities associated with his disease.
CASE PRESENTATION WITH IMAGING
5 year old African American male presented to the hospital with chief complaint of worsening scrotal swelling and chronic diarrhea. Toddler was born premature at 35 weeks, SGA with 4Ibs 7oz. He was hospitalized initially at two months of age for GERD and neutropenia which self-resolved. Throughout, he was hospitalized due to multiple infections ranging from pneumonia to ear infections to preseptal cellulitis. At three years of age, he developed profuse diarrhea with foul smelling which consisted of greater than 10 stools per day with pallor and fatigue. Work up was significant for a negative stool culture, a positive guaiac, negative stool fat, elevated fecal calprotectin, normal stool apha-1 antitrypsin, normal PH, negative tTG Ab, and normal trypsin.
In addition, he was also noted to have swelling of his scrotum and was admitted multiple times for “recurrent cellulitis” despite being on antibiotics. Notably, with his swelling, his skin would get irritated and red over the scrotum, gluteal folds as well as perianal region. His swelling of the scrotum would improve but symptoms would recur after 1-2 weeks post treatment. At presentation, toddler had stable vital signs being afebrile with a pulse rate of 100, with respiratory rate of 26, and blood pressure of 94/61. Weight was noted to be 20kg (38% for his age). Physical examination was significant for dysmorphic facial features with saddle nose, hypertelorism, misaligned teeth and high arched palate. Genital exam showed a mild scrotal swelling with thick lichenified skin over the scrotum, perianal area. Rectal exam was reassuring with normal anal tone. Laboratory findings such as CRP and ESR were mildly elevated. Ultrasound of the scrotum was done which showed diffuse severe scrotal edema with associated of hyperemia, with normal size of the testis without any hydrocele or extra-testicular mass. Fecal calprotectin was found to be 1300 which prompted further investigation. Upper endoscopy and colonoscopy showed pancolitis with serpiginous ulcers as well as pseudopolyps concerning for early onset Crohn’s disease. Since then, he has been treated with sulfasalazine, prednisone taper and three weeks course of flagyl which improved his scrotal swelling.
Due to significant developmental delay, dysmorphic features and multiple co morbidities such as Crohn’s disease, iron deficiency anemia, Whole Exome Sequencing was performed. He was identified to have a homozygous pathogenic variant in the PEPD gene which was associated with autosomal recessive Prolidase deficiency. Though, he has multiple features that are associated with Prolidase deficiency such as dysmorphic features, developmental delay, chronic anemia, recurrent infections and skin lesions; there has been little information in the literature with regards to the association between Prolidase deficiency and Crohn’s disease.
In addition, he was seen by Pediatric Dermatologist for his scrotal swelling and new onset of non-healing left leg ulceration. His skin ulceration continued to worsen with new wounds spreading to lower extremities bilaterally consistent Prolidase deficiency(Figure 1).
In addition, he was also noted to have swelling of his scrotum and was admitted multiple times for “recurrent cellulitis” despite being on antibiotics. Notably, with his swelling, his skin would get irritated and red over the scrotum, gluteal folds as well as perianal region. His swelling of the scrotum would improve but symptoms would recur after 1-2 weeks post treatment. At presentation, toddler had stable vital signs being afebrile with a pulse rate of 100, with respiratory rate of 26, and blood pressure of 94/61. Weight was noted to be 20kg (38% for his age). Physical examination was significant for dysmorphic facial features with saddle nose, hypertelorism, misaligned teeth and high arched palate. Genital exam showed a mild scrotal swelling with thick lichenified skin over the scrotum, perianal area. Rectal exam was reassuring with normal anal tone. Laboratory findings such as CRP and ESR were mildly elevated. Ultrasound of the scrotum was done which showed diffuse severe scrotal edema with associated of hyperemia, with normal size of the testis without any hydrocele or extra-testicular mass. Fecal calprotectin was found to be 1300 which prompted further investigation. Upper endoscopy and colonoscopy showed pancolitis with serpiginous ulcers as well as pseudopolyps concerning for early onset Crohn’s disease. Since then, he has been treated with sulfasalazine, prednisone taper and three weeks course of flagyl which improved his scrotal swelling.
Due to significant developmental delay, dysmorphic features and multiple co morbidities such as Crohn’s disease, iron deficiency anemia, Whole Exome Sequencing was performed. He was identified to have a homozygous pathogenic variant in the PEPD gene which was associated with autosomal recessive Prolidase deficiency. Though, he has multiple features that are associated with Prolidase deficiency such as dysmorphic features, developmental delay, chronic anemia, recurrent infections and skin lesions; there has been little information in the literature with regards to the association between Prolidase deficiency and Crohn’s disease.
In addition, he was seen by Pediatric Dermatologist for his scrotal swelling and new onset of non-healing left leg ulceration. His skin ulceration continued to worsen with new wounds spreading to lower extremities bilaterally consistent Prolidase deficiency(Figure 1).
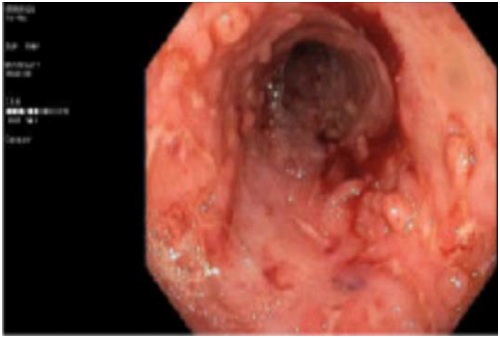 Figure 1: Colonoscopy results showing pancolitis with serpiginous ulcers as well as pseudopolyps.
Figure 1: Colonoscopy results showing pancolitis with serpiginous ulcers as well as pseudopolyps.DISCUSSION
Crohn’s disease is a granulomatous disorder with an unknown etiology that presents in adolescent and young adults with subtle symptoms such as diarrhea, weight loss, anorexia and abdominal pain. Though, uncommon, extra-intestinal manifestation can precede typical bowel findings of Crohn’s and may complicate the disease process and can make difficult for clinicians to diagnose. These extra-intestinal manifestations include but not limited to arthritis, iritis, and uveitis, cutaneous lesions such as erythema nodosum and pyoderma gangrenosum[10]. Skin lesions can occur in 14-44% of patients with Crohn’s disease [8]; these lesions are seen commonly in patient with colonic involvement and unrelated to the activity of bowel disease [10]. Cutaneous Crohn’s disease can be divided into two categories: genital vs.non-genital. It is estimated that about two thirds of children with cutaneous Crohn’s disease have a genital involvement. Classically, these cutaneous lesions present as subcutaneous nodules or erythematous plaques or secondary ulcers [10].
Our patient presented with cellulitis and edema of the scrotum which is usually unusual for cutaneous Crohn’s disease. Management of cutaneous Crohn’s disease is difficult and refractory. However, significant studies have shown medications such as metronidazole, steroids and sulfasalazine are effective. After treatment our patient with metronidazole, steroids and sulfasalazine, his cutaneous swelling of the scrotum improved. In addition, our patient later presented with cutaneous ulcers typical of prolidase deficiency.
Prolidase deficiency disorder is a very rare disorder that presents with facial anomaly, saddle nose, frontal bossing, hypertelorisim and mandibular protrusion. Most common manifestation of Prolidase deficiency is involvement of the skin with classic irregularly shaped ulcers with prominent of granulation tissue involving lower extremities. These ulcers heal with scars and are pitting in nature. Patient with PD have increased incidence of infection due to splenic dysfunction caused by amyloid deposition [7]. Due to deficiency of the prolidase enzyme, excess of increase urinary dipeptides such as proline and hydroxyl proline cause defective collagen synthesis and impaired wound healing.
Association of prolidase deficiency and Lupus erythematous has been well described in literature suggesting that prolidase deficiency might be a risk factor for development of SLE [8]. However, only one case report has been found with association of IBD and prolidase deficiency. We report our case to emphasize and begs the question about relation between prolidase deficiency and early onset Crohn’s disease. When considering differential diagnosis for scrotal swelling, facial anomalies, recurrent skin ulcers, and developmental delay with associated chronic diarrhea related to Crohn’s disease, prolidase deficiency should be considered.
Our patient presented with cellulitis and edema of the scrotum which is usually unusual for cutaneous Crohn’s disease. Management of cutaneous Crohn’s disease is difficult and refractory. However, significant studies have shown medications such as metronidazole, steroids and sulfasalazine are effective. After treatment our patient with metronidazole, steroids and sulfasalazine, his cutaneous swelling of the scrotum improved. In addition, our patient later presented with cutaneous ulcers typical of prolidase deficiency.
Prolidase deficiency disorder is a very rare disorder that presents with facial anomaly, saddle nose, frontal bossing, hypertelorisim and mandibular protrusion. Most common manifestation of Prolidase deficiency is involvement of the skin with classic irregularly shaped ulcers with prominent of granulation tissue involving lower extremities. These ulcers heal with scars and are pitting in nature. Patient with PD have increased incidence of infection due to splenic dysfunction caused by amyloid deposition [7]. Due to deficiency of the prolidase enzyme, excess of increase urinary dipeptides such as proline and hydroxyl proline cause defective collagen synthesis and impaired wound healing.
Association of prolidase deficiency and Lupus erythematous has been well described in literature suggesting that prolidase deficiency might be a risk factor for development of SLE [8]. However, only one case report has been found with association of IBD and prolidase deficiency. We report our case to emphasize and begs the question about relation between prolidase deficiency and early onset Crohn’s disease. When considering differential diagnosis for scrotal swelling, facial anomalies, recurrent skin ulcers, and developmental delay with associated chronic diarrhea related to Crohn’s disease, prolidase deficiency should be considered.
CONCLUSION
This case illustrates a young child with unusual presentation of cutaneous Crohn’s disease along with very early-onset Crohn’s disease whose hospital course was complicated with Prolidase deficiency. Emerging data suggest that very early-onset IBD will have more severe symptoms with unique phenotype and genotype; comprehensive investigation must be done as VOD-Crohn’s can predispose to other genetic disorder.
REFERENCES
- Goodman SI, Solomons CC, Muschenheim F, McIntyre CA, Miles B, et al. (1968) A syndrome resembling lathyrism associated with iminodipeptiduria. Am J Med 45: 152-159.
- Kurien BT, D'Sousa A, Bruner BF, Gross T, James JA, et al. (2013) Prolidase deficiency breaks tolerance to lupus-associated antigens. Int J Rheum Dis 16: 674-680.
- Kuloglu Z, Kansu A, Demir A, Yaman A, Serwas N, et al. (2015) Inflammatory bowel disease-like phenotype in a young girl with prolidase deficiency: a new spectrum of clinical manifestation. Genet Couns 26: 205-211.
- Glick SR, Carvalho RS (2011) Inflammatory bowel disease. Pediatr Rev 32: 14-24.
- Benchimol EI, Guttmann A, Griffiths AM, Rabeneck L, Mack DR, et al. (2009) Increasing incidence of paediatric inflammatory bowel disease in Ontario, Canada: evidence from health administrative data. Gut 58: 1490-1497.
- Bissonnette R, Friedmann D, Giroux JM, Dolenga M, Hechtman P, et al. (1993) Prolidase deficiency: a multisystemic hereditary disorder. J Am AcadDermatol 29: 818-821.
- ButbulAviel Y, Mandel H, AvitanHersh E, Bergman R, Adiv OE, et al. (2012) Prolidase deficiency associated with systemic lupus erythematosus (SLE): single site experience and literature review. PediatrRheumatol Online J 10:18.
- Kelsen JR, Dawany N, Moran CJ, Petersen BS, Sarmady M, et al. (2015) Exome sequencing analysis reveals variants in primary immunodeficiency genes in patients with very early onset inflammatory bowel disease. Gastroenterology 149: 1415-1424.
- Rankin GB, Watts HD, Melnyk CS, Kelley Jr ML (1979) National cooperative Crohn's disease study: extraintestinal manifestations and perianal complication. Gastroenterology 77: 914-920.
- Shrinath M, Walter JH, Haeney M, Couriel JM, Lewis MA, et al. (1997) Prolidase deficiency and systemic lupus erythematosus. Arch Dis Child 76: 441-444.
Citation: Rizvi SA, Elder M and Beasley G(2019) A Novel Manifestation of Prolidase Deficiency in a Toddler Diagnosed with Very-Early Onset Crohn’s Disease. J Gastroenterol Hepatology Res S1: 001.
Copyright: © 2019 S. Ahsan Rizvi, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Journal Highlights
© 2026, Copyrights Herald Scholarly Open Access. All Rights Reserved!