A Novel Splice-Site Mutation of the Gk Gene in a Chinese Patient with Isolate Glycerol Kinase Deficiency
*Corresponding Author(s):
Lingqian WuCenter For Medical Genetics, School Of Life Sciences, Hunan Jiahui Genetics Hospital, Central South University, 110 Xiangya Road, Changsha, Hunan 410078, China
Tel:+86 73184805252,
Fax:+86 731 84478152
Email:wulingqian@sklmg.edu.cn
Desheng Liang
Center For Medical Genetics, School Of Life Sciences, Hunan Jiahui Genetics Hospital, Central South University, 110 Xiangya Road, Changsha, Hunan 410078, China
Tel:+86 73184805252,
Fax:+86 73184478152
Email:liangdesheng@sklmg.edu.cn
Abstract
What is already known on this topic?
Keywords
INTRODUCTION
Glycerol Kinase Deficiency (GKD) [OMIM 307030] is a rare X-linked recessive metabolic disorder, which is characterized biochemically by hyperglycerolemia and glyceroluria [1]. The clinical spectrum varies from asymptomatic and Central Nervous System (CNS) deterioration to severe developmental delay, and is found to be related with GK gene mutation. The gene (GK) is located on Xp21 and encodes a key enzyme in regulation of glycerol uptake and metabolism that catalyzes the phosphorylation of glycerol [2].
GKD can be categorized into three types: Infantile GKD, Juvenile GKD and Adult GKD. The Infantile GKD, also called the complex GKD or Xp21 contiguous gene deletion syndrome, is the most common type of GK mutation, which is caused by a deletion of GK gene or the neighboring genes, the clinical manifestations depends on the deletion of the genes [3]. The juvenile GKD is called symptomatic GKD, with symptoms like episodic vomiting, acidemia and Central Nervous System (CNS) deterioration. The adult GKD is called asymptomatic GKD, which detected incidentally with pseudohypertriglyceridemia. The juvenile and adult GKD are also called isolated GKD, which mainly caused by point mutations in GK gene.
In this case report, a Chinese patient clinically diagnosed with isolated glycerol kinase deficiency is detected to have a novel splice-site mutation in the GK gene, which leads to a skip of exon 17.
CASE REPORT
General information and clinical manifestations
Laboratory tests
Molecular studies
Mutation screening of the GK gene by Sanger sequencing:
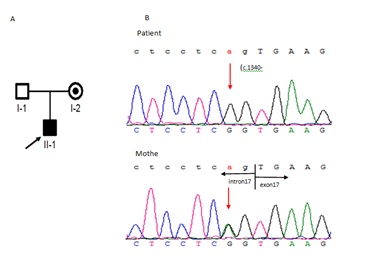
We identified a split-site mutation (c.1340 -2A>G) in the GK gene (NM_000167), his mother was heterozygous (Figure 1) for the mutation, and it had not been previously reported in the Human Gene Mutation Database (HGMD), dbSNP144, or Exome Variant Server (EVS). To investigate the effect of this splice-site mutation on RNA species, we performed Reverse transcription-PCR on RNA from the patient’s peripheral blood samples.
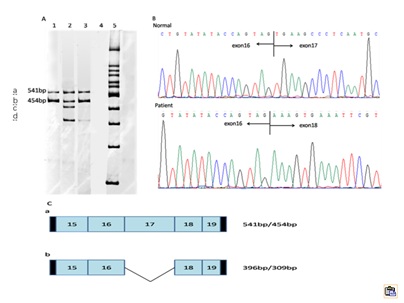
Mutation analysis of cDNA by Reverse transcription PCR:

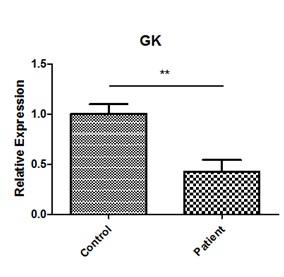
Analysis of GK expression by real-time quantitative PCR:
DISCUSSION
Currently, there are about 34 mutations in GK gene (NM_000167) have been reported in patients with GKD (HGMD Professional 2017.1), including missense/nonsense mutations (60-61%), splice-site mutations (21-22%), deletion mutations(15-16%) and insertion mutations (3-4%).
The GK gene (NM_000167) contains 19 exons and yield 1575bp transcript. We identified a patient with isolated GKD due to splice-site mutation (c.1340 -2 A>G). RT-PCR analysis of the patient’ s peripheral blood lymphocyte cell revealed that there were four fragments, two wild-type GK mRNA, and two mutant mRNA. The mutation created a cryptic splice acceptor site, resulting in alternative splicing of the transcript with a small amount of correctly spliced GK mRNA. Likewise, we found that there were normal splicing in the splice-site mutations reported previous [4,5]. Sequencing the fragments, we found that the mutation caused a skipping of the whole exon17, leading a frame shift mutant of p. Val446Glufs*48. Then, we did the real-time PCR, and found that the GK mRNA in patient was decreased by 59.2%, compared to the control. So, we can get a conclusion that the splice-site mutation (c.1340 -2 A>G) of GK may be the cause of the patient.
Previous studies had also reported the deletion of exon17 in two isolated GKD patients, one presented psychomotor, growth retardation and multiple fractures from minor trauma, which couldn’t be prevented by vitamin D or phosphate supplements, and another one manifested pale, somnolent, hyperventilate and severe abdominal pains [6,7]. It’s suggested that the deletion of exon17 is strongly associated with the incidence of GKD.
Interestingly, most of the patients with split-site mutation reported previously presented with asymptomatic form of GKD [4,5,7-9]. Only one patient from the literature (IVS9-1G>T) who had a history of metabolic and CNS crises at the age 6 years and 3 months, indicating the symptomatic form of GKD, but he was uneventful in the neonatal period and early childhood [10].
Previous studies showed that there was no genotype-phenotype correlation in GKD [1,11,12]. Our patient presented no symptoms, excepting no weight gain. The probable causes may be as followings: First, it may be related to age, just like the patient reported by Hellerud et al. [10], the patient didn’t present symptoms at an early age. Second, the patient may still have part of normal mRNA, so the clinical symptom is mild. Third, it may be related to Nonsense Mediated Decay (NMD). Zhang et al., found that aberrant mRNA generated by splice site mutations of the GK gene can be eliminated by NMD in the asymptomatic form of GKD [9]. In contrary, there was no NMD in our study, and reviewed the other split-site mutations previously reported, no NMD was found too [4,5]. Fourth, some studies demonstrated that glycerolutilization by the cells (especially, hepatocytes) is rate-limited by the membrane permeation step, rather than the GK step [13-15]. AQP9, an aquaglyceroporin, has been reported to be of great significance in glycerol membrane permeation step [13-15]. Expression of AQP9 is influenced by gender and disease state. In human, despite exhibiting similar hepatic AQP9 protein, women manifest lower hepatocyte glycerol permeability compared to men [16]. In obese subjects with Non-Alcoholic Fatty Liver Disease (NAFLD), the hepatic expression of AQP9 was decreased [16]. In the rodent, female rats have lower levels of hepatic AQP9 protein compared with males in both fed and fasting conditions. The mice model of NAFLD, demonstrated decreased hepatocyte AQP9 and glycerol permeability correlated with increased plasma glycerol [16], andAQP9-/- mice exhibited a marked increase in plasma glycerol and triglyceride levels compared with AQP9+/- mice, revealing a deficient glycerol metabolism [17]. Therefore, in addition to the GK mutation that causes a high serum triglyceride, the possible role of AQP9 needs to be emphasized, but this still needs to be further studied in GKD patients.
Glycerol kinase deficiency has a suspected risk for pancreatitis and coronary heart disease, and there is no effective way to cure GKD, but there may be some treatment, such as aggressive Therapeutic Lifestyle and combination lipid-altering drug, to reduce the serum glycerol.
CONCLUSION
We identified a novel splice-site mutation c.1340 -2 A>G in the GK gene, which was most likely responsible for this patient’s pathogenic cause. This detection offered an important insight into the pathogenic mutation spectrum of glycerol kinase deficiency and could help in clinical diagnosis and genetic counseling of patients with glycerol kinase deficiency.
CONFLICT OF INTEREST
There is no conflict of interest.
ACKNOWLEDGMENT
We thank the patient’s family for their help and support. This study was supported by the National Natural Science Foundation of China (Grant number 81501268, 81571450) and the Key Research and Development Program of Hunan Province (Grant number 2016WK2029).
REFERENCES
- Sjarif DR, Ploos van Amstel JK, Duran M, Beemer FA, Poll-The BT (2000) Isolated and contiguous glycerol kinase gene disorders: a review. J Inherit Metab Dis 23: 529-547.
- Stanczak CM, Chen Z, Zhang YH, Nelson SF, McCabe ER (2007) Deletion mapping in Xp21 for patients with complex glycerol kinase deficiency using SNP mapping arrays. Hum Mutat 28: 235-242.
- Sevim U, Fatma D, Ihsan E, Gulay C, Nevin B (2011) A neonate with contiguous deletion syndrome in XP21. J Pediatr Endocrinol Metab 24: 1095-1098.
- Sargent CA, Kidd A, Moore S, Dean J, Besley GT, et al. (2000) Five cases of isolated glycerol kinase deficiency, including two families: failure to find genotype: phenotype correlation. J Med Genet 37: 434-441.
- Sjarif DR, Hellerud C, van Amstel JK, Kleijer WJ, Sperl W, et al. (2004) Glycerol kinase deficiency: residual activity explained by reduced transcription and enzyme conformation. Eur J Hum Genet 12: 424-432.
- Hellerud C, Wramner N, Erikson A, Johansson A, Samuelson G, et al. (2004) Glycerol kinase deficiency: follow-up during 20 years, genetics, biochemistry and prognosis. Acta Paediatr 93: 911-921.
- Walker AP, Muscatelli F, Stafford AN, Chelly J, Dahl N, et al. (1996) Mutations and phenotype in isolated glycerol kinase deficiency. Am J Hum Genet 58: 1205-1211.
- Hellerud C, Burlina A, Gabelli C, Ellis JR, Nyholm PG, et al. (2003) Glycerol metabolism and the determination of triglycerides--clinical, biochemical and molecular findings in six subjects. Clin Chem Lab Med 41: 46-55.
- Zhang YH, Huang BL, Jialal I, Northrup H, McCabe ER, et al. (2006) Asymptomatic isolated human glycerol kinase deficiency associated with splice-site mutations and nonsense-mediated decay of mutant RNA. Pediatr Res 59: 590-592.
- Hellerud C, Adamowicz M, Jurkiewicz D, Taybert J, Kubalska J, et al. (2003) Clinical heterogeneity and molecular findings in five polish patients with glycerol kinase deficiency: investigation of two splice site mutations with computerized splice junction analysis and Xp21 gene-specific mRNA analysis. Mol Genet Metab 79: 149-159.
- Dipple KM, Zhang YH, Huang BL, McCabe LL, Dallongeville J, et al. (2001) Glycerol kinase deficiency: evidence for complexity in a single gene disorder. Hum Genet 109: 55-62.
- Rahib L, MacLennan NK, Horvath S, Liao JC, Dipple KM (2007) Glycerol kinase deficiency alters expression of genes involved in lipid metabolism, carbohydrate metabolism, and insulin signaling. Eur J Hum Genet 15: 646-657.
- Calamita G, Gena P, Ferri D, Rosito A, Rojek A, et al. (2012) Biophysical assessment of aquaporin-9 as principal facilitative pathway in mouse liver import of glucogenetic glycerol. Biol Cell 104: 342-351.
- Li CC, Lin EC (1983) Glycerol transport and phosphorylation by rat hepatocytes. J Cell Physiol 117: 230-234.
- Rodriguez A, Catalán V, Gómez-Ambrosi J, García-Navarro S, Rotellar F, et al. (2011) Insulin- and leptin-mediated control of aquaglyceroporins in human adipocytes and hepatocytes is mediated via the PI3K/Akt/mTOR signaling cascade. J Clin Endocrinol Metab 96: 586-597.
- Rodriguez A, Marinelli RA, Tesse A, Frühbeck G, Calamita G (2015) Sexual Dimorphism of Adipose and Hepatic Aquaglyceroporins in Health and Metabolic Disorders. Front Endocrinol (Lausanne) 6: 171.
- Rojek AM, Skowronski MT, Füchtbauer EM, Füchtbauer AC, Fenton RA, et al. (2007) Defective glycerol metabolism in Aquaporin 9 (AQP9) knockout mice. Proc Natl Acad Sci U S A 104: 3609-3614.
Citation: Zeng L, Mei L, Tan H, Li H, Lv W, et al. (2017) A Novel Splice-Site Mutation of the Gk Gene in a Chinese Patient with Isolate Glycerol Kinase Deficiency. J Hum Endocrinol 2: 011.
Copyright: © 2017 Lingqian Wu, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.