A Rare Mutation in Noonan Syndrome
*Corresponding Author(s):
Vasco CarvalhoNeonatal Intensive Care Unit, Department Of Pediatrics, Hospital Of Braga, Braga, Portugal
Tel:+351 967109619,
Email:vascofonsecacarvalho@gmail.com
Abstract
Introduction
Noonan Syndrome (NS) is a genetic disorder mainly characterized by short stature, distinctive facial features, congenital heart defects, cardiomyopathy and an increased risk to develop tumors in childhood. The incidence is estimated to be between 1:1000 and 1:2500 live births. Mutations in PTPN11 (12q24.13) are seen in 50% of cases.
Description of case
Infant girl, born to healthy non-consanguineous parents with an unremarkable family history. Fetal ultrasounds revealed bilateral cervical cysts at first trimester, bilateral hydronephrosis and polyhydramnios at second and third trimesters. Eutocic delivery at 38th week gestation with a good transition to extra-uterine life and appropriate somatometry for gestational age.
Admitted to NICU on the third day of life with transient episodes of cyanosis. Echocardiogram showed pulmonary stenosis and chest radiography was normal. She was discharged at day six of life for ambulatory surveillance. In the evaluations of the first months, we observed a faster proliferation cervical hemangioma and some other phenotypic findings: Hypertelorism, epicanthic folds, high nasal bridge, webbed neck and low-set ears. Presumptive diagnosis of NS was done. Propranolol treatment was started for the cervical hemangioma. Genetic study revealed a rare mutation in BRAF gene, variant c1789>G p, with pathogenic value. Crossing of percentiles in length was found, being in P3. Nowadays, with 17 months old, she maintains a multidisciplinary approach.
Conclusion
Being a rare mutation of NS and the overlapping features with other syndromes, it is necessary to have a high suspicion level, beginning in the prenatal phase. In this clinical case, the recognition of typical features allows the diligent follow up and consequent diagnosis of a disease.
Keywords
BRAF; Congenital heart defects; Noonan syndrome; Phenotype-genotype correlations; RAS-MAPK pathway
INTRODUCTION
Noonan Syndrome (NS) is a genetic disorder. It has been described by Jacqueline Anne Noonan and Dorothy Ehmke in 1963 and its incidence range lies between 1:1,000 to 1:2,500 live births and it affects equally males and females [1].
It is genetically heterogeneous with at least eight causative genes acting in the RAS/MAPK signaling pathway [2]. Though most of the cases are autosomally inherited some cases may be sporadic [3].
The pathophysiology of Noonan syndrome is not fully understood. The most common gene associated with NS is PTPN11 which accounts for approximately half of all cases. Other disease-causing genes (SOS1, RAF1, KRAS) have been identified. All 4 genes are part of the RAS/RAF/MEK/ERK signal transduction pathway, which is an important regulator of cell growth. Mutations in the RASMAPK signaling pathway are typically responsible for Noonan syndrome [3].
Although it is clinically heterogeneous and can present at any age, NS is diagnosed on clinical grounds by observation of key features [4].
The cardinal features of NS include unusual facies with typical triangular craniofacial dysmorphism combining hypertelorism, a broad forehead, down-slanting palpebral fissures, low set ears posteriorly rotated, a short and webbed neck. Skin and hair abnormalities can also be found, with wispy hair and cutaneous hemangioma as the most common findings [1,3].
More than 80 percent of patients have cardiac involvement, most often Pulmonary valve Stenosis (PS) with dysplastic pulmonary valve. PS can be associated with hypertrophic cardiomyopathy [1].
Skeletal, neurologic, genitourinary, lymphatic, skin, and ocular findings may be present to varying degrees. This syndrome courses in majority of times with short stature. Skeletal deformities (extremities, chest or spinal anomalies) are less frequencies. Approximately 25% of individuals with Noonan syndrome have mental retardation.
Ophthalmologic problems can also be present. There is a high incidence of lesions of the eye, usually hypertelorism, down-slanting palpebral fissures, epicanthic folds, ptosis, refractive errors, strabismus, amblyopia, nystagmus, and rarely cataract, colobomas, and keratoconus [5].
Hearing difficulties are frequent too. Cryptorchidism occurs in 80% of male cases. Renal malformations, coagulation defects and hypogonadism can be founded [3]. Oral and dental issues of NS include a high arched palate, dental malocclusion, ectopically positioned teeth, micrognathia, articulation difficulties, delayed tooth eruption, dental erosion, and multiple dental caries [6].
CASE REPORT
This infant girl was born to healthy non-consanguineous parents. She had a healthy 3-year-old sister. The family history was unremarkable. It was a pregnancy complicated by gestational diabetes treated with diet. The fetal ultrasounds revealed bilateral cervical cysts at first trimester, bilateral hydronephrosis (right 7,5 mm and left 10,5 mm) and polyhydramnios at second and third trimesters.
She was eutocic delivered at 38th week gestation. The Apgar scores were 10/10/10 at one, five and ten minutes respectively. The body weight was 3825 grams (P90), height 49 cm (P50), head circumference 35,5 cm (P90), appropriate for gestational age. The screening for congenital heart disease and hearing loss were normal.
She was admitted on day 3 of life in the Neonatal Department due to cyanosis episodes with pulse oximetry levels of 91-93% and systolic murmur. Echocardiogram showed pulmonary stenosis with pulmonary arterial pressure of 45 mmHg. Chest radiogram was normal. She was discharged at day six of life for ambulatory surveillance.
On postnatal renal-pelvic ultrasound was observed a left pelvis dilatation of 6mm. The transfontanellar ultrasound was normal. In the evaluations of the first months, we identified a cervical hemangioma (Figure 1) in a faster proliferation phase and other phenotypic findings: Hypertelorism, epicanthic folds, flat nasal bridge, broad forehead, cupid bow appearance of upper lip, wispy hair and low-set ears. A crossing of percentiles in length was found, being in P3 at 2-months-old. Presumptive diagnosis of NS was done. Propranolol treatment started for the cervical hemangioma.
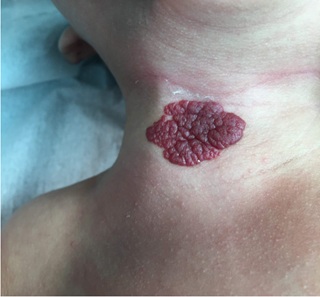 Figure 1: Cervical hemangioma.
Figure 1: Cervical hemangioma.
Genetic study revealed a rare mutation in BRAF gene, variant c1789>G p, with pathogenic value. Crossing of percentiles in length was found, being in P3. Now, with a 17-month-old, she maintains a multidisciplinary approach. She maintains the facial features (Figures 2 & 3), the length at P3 and normal psychomotor development.
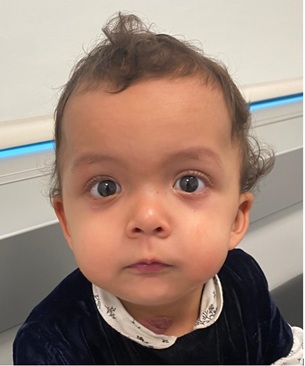 Figure 2: Frontal photography showing typical features of Noonan Syndrome like hypertelorism, flat nasal bridge, broad forehead, cupid bow appearance of upper lip and wispy hair.
Figure 2: Frontal photography showing typical features of Noonan Syndrome like hypertelorism, flat nasal bridge, broad forehead, cupid bow appearance of upper lip and wispy hair.
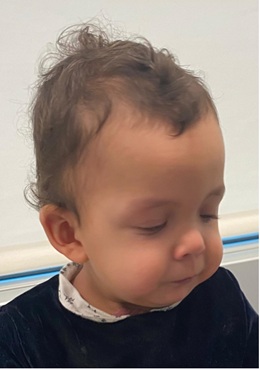 Figure 3: Lateral photography highlighting typical features of Noonan Syndrome like low set ears.
Figure 3: Lateral photography highlighting typical features of Noonan Syndrome like low set ears.
DISCUSSION
The NS has overlapping features with other syndromes, so it’s essentially a high suspicion level, beginning in the prenatal phase. Sarkozy et al. [8], identified heterozygous de novo mutations in the BRAF gene in 5 of 270 patients with NS. Common clinical features of those 5-patient included poor growth, variable feeding difficulties, short stature, mild cognitive defects, and hypotonia. Dysmorphic facial features included prominent forehead, hypertelorism and low-set ears with thickened helices and wispy hair. Two patients had congenital cardiac defects, pulmonary stenosis and atrial septal defect, respectively and 3 had hyperpigmented cutaneous lesions. The NS with mutation in BRAF gene (MIM #613706), described by Sarkozy et al and reported in this case is rare, found in only 2% of all NS [4]. BRAF mutation was also described in patients with sporadic pyogenic granuloma and pyogenic granuloma associated with port wine stains [9].
In this clinical case, given the bilateral cervical cysts associated with bilateral hydronephrosis and polyhydramnios at second and third trimesters in the prenatal ultrasounds, the suspicion of a syndrome could have been put. The first manifestation of this girl, cyanosis, at day 3 of life, allows the detection of PS. Noonan syndrome is one of the most common genetic disorders associated with congenital heart defects, being second only to Down syndrome [2]. In this case, it was essential the detection of pulmonar stenosis that associated with typical phenotypic characteristics, raised the hypothesis of NS.
Another fact that made the clinical diagnosis more likely was the cross of percentiles in length to P3, once short stature is present in the majority cases of NS. This way, the recognition of typical features allows the diligent follow up and consequent diagnosis of a disease. The children with Noonan syndrome usually have a wide array of health problems, making it important for all practitioners to be aware of the child’s special care needs. Multidisciplinary treatment is the key to success in managing children with syndromes, like in this girl.
DECLARATION OF INTEREST
The Authors declare that there is no conflict of interest.
REFERENCES
- Poaty H, Cardorelle AM, Moukouma C, Mouko A (2017) Clinical diagnosis of noonan syndrome and brief review of literature. Ann Med Health Sci Res 7: 76-79.
- Setty HSN, Shankar S, Patil R, Jadhav S, Yeriswmy MC, et al. (2020) Combined cardiac anomalies in Noonan syndrome: A case report. International Journal of Surgery Case Reports 72: 32-36.
- Kouz K, Lissewski C, Spranger S, Diana M, Angelika R, et al. (2016) Genotype and phenotype in patients with Noonan syndrome and a RIT1 mutation. Genet Med 18: 1226-1234.
- Allanson JE, Roberts AE (2001) Noonan Syndrome. GeneReviews®, Seattle, USA.
- Vujanovic M, Stankovic-Babic G, Cekic S (2014) Noonan Syndrome - case report. Acta Medica Medianae 53: 54-56.
- Hwang I, Lee Y, Sim D, Mah Y (2018) Oral features in a child with noonan syndrome: A case report. J Korean Acad Pediatr Dent 45: 115-122.
- Gelb BD, Tartaglia M (1993) Noonan Syndrome with Multiple Lentigines. GeneReviews®, University of Washington, Seattle, USA.
- Sarkozy A, Carta C, Moretti S, Zampino G, Digilio MC, et al. (2009) Germline BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: Molecular diversity and associated phenotypic spectrum. Hum Mutat 30: 695-702.
- Groesser L, Peterhof E, Evert M, Landthaler M, Berneburg M, et al. (2016) BRAF and RAS mutations in sporadic and secondary pyogenic granuloma. J Invest Dermatol 136: 481-486.
Citation: Carvalho V, Vilaça J, Leite L, Rocha MG, Sousa G, et al. (2020) A Rare Mutation in Noonan Syndrome. J Neonatol Clin Pediatr 7: 060.
Copyright: © 2020 Vasco Carvalho, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.