A Solitary Infantile Myofibroma – Case Report
*Corresponding Author(s):
Sofia Brandão MirandaPediatric Department, Hospital De Braga, Braga, Portugal
Tel:+351 926702396,
Email:asofiabmiranda@gmail.com
Maria Ventura Nogueira
Pediatric Department, Hospital De Braga, Braga, Portugal
Tel:+351 918655095,
Email:mj.ventura.nogueira@gmail.com
Abstract
Infantile Myofibroma (IM) is a rare, benign tumor that affects infants and young children, presenting as a solitary or multicentric nodular mass involving the skin, muscles, bones, and/or visceral organs. A 6-year-old girl was referred for a Pediatric Oncology appointment for a right temporal mass that had been noticed 20 days earlier. Ultrasound, CT, and MRI revealed a lenticular structure centered in bone, with a soft tissue component, raising the possibility of Langerhans cell histiocytosis. A biopsy was performed and histology revealed a mesenchymal neoplasm consistent with IM. Follow-up over the next 24 months showed no signs of recurrence or malignant transformation. Surgery is typically performed to prevent complications or improve prognosis, with a 10% rate of recurrence. Chemotherapy may be used in cases of involvement of internal organs, unsuccessful surgery, or in unresectable lesions. Histopathology is the gold standard for diagnosis, but due to the rarity of the disease, there are no standardized treatment protocols.
Keywords
Case report; Infantile myofibroma; Langerhans cell histiocytosis; Pediatric patient; Solitary mass
Introduction
Infantile myofibroma is a rare, benign tumor that affects infants and young children. It presents as a solitary or multicentric nodular mass involving the skin, muscles, bones, and/or visceral organs. Its clinical presentation may be similar to other more common conditions, making histopathology the gold standard for diagnosis [1].
Case Report
6-year-old girl was referred to a Pediatric Oncology appointment with a right temporal mass that her mother had noticed 20 days earlier. There was no history of trauma or pain, or any changes in weight loss, appetite and urinary or fluid intake. On physical examination, she was in good general condition. Palpation of the skull revealed a well-defined, hard swelling on the right temporal region, measuring 20 mm, without any inflammatory signs (Figure 1). No other swellings or palpable lymph nodes were noted.
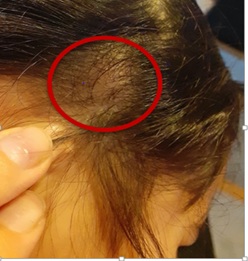 Figure 1: Clinical photograph showing the right temporal mass.
Figure 1: Clinical photograph showing the right temporal mass.
An ultrasound of the mass had already been performed, revealing a lenticular structure measuring 16x5 mm that appeared to be centered in bone, with a soft tissue component. This raised the possibility of Langerhans cell histiocytosis and suggested further characterization with a Computed Tomography (CT) scan. Cranial CT and Magnetic Resonance Imaging (MRI) descriptions maintained the same diagnostic suspicion (Figure 2). Analytically, blood count, urinalysis, ionogram, urinary and blood osmolality, hepatic and renal function, and sedimentation rate were normal. Noteworthy only lactate dehydrogenase of 265 U/L. Full-body bone x-ray did not show other lesions and pulmonary radiograph and abdominal ultrasound were performed with no relevant findings. Positron Emission Tomography (PET) scan was performed with no significant or anomalous 2-deoxy-2-[fluorine-18] fluoro-D-glucose [2].
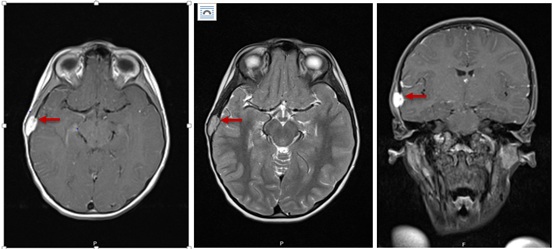 Figure 2: Cranial Magnetic Resonance Imaging (MRI) scans showing the right temporal mass.
Figure 2: Cranial Magnetic Resonance Imaging (MRI) scans showing the right temporal mass.
In a multidisciplinary appointment it was decided to perform a biopsy to better characterize the lesion.The histology revealed a mesenchymal neoplasm, moderately cellular, composed of cells with an oval, rounded or stellate nucleus, slightly hyperchromatic and with scanty and bipolar of moderate eosinophilic cytoplasm. The cells were arranged in bundles of varying size and orientation within a partly lax and partly hyalinized streak, mixed with a discrete lymphocytic inflammatory infiltrate. Light microscopy and immunohistochemistry showed diffuse positivity for α-smooth muscle actin and calponin, multifocal for CD99 and focal for caldesmon. In immunophenotyping it was not detected CD1a+ cCD68+ cells, typical from Langerhans cells. The aspects described, framed in the clinical and imaging context, favor the diagnosis of Solitary Infantile Myofibroma (IM).
It was decided to maintain clinical and imaging follow-up with cranial MRI, abdominal and pelvic MRI/ultrasound. After 24 months of follow-up, there were no signs of recurrence or malignant transformation
IM is a rare, benign soft tissue tumor that affects infants and young children. It is characterized by the development of a solitary (75% of cases) or multicentric nodular mass involving the skin, striated muscles, bones, and in exceptional cases, visceral organs. In its solitary form, the nodule is usually found in the head, neck, or trunk areas. The exact incidence is unknown, due to undiagnosed or misdiagnosed cases, but it is estimated to be between 1 in 150,000 to 400,000 live births. Most of these tumors are sporadic and isolated, but rare familial cases have been described [3].
Differential diagnoses for this condition include hemangioma, lymphangioma, neurofibroma, infantile fibrosarcoma, Langerhans cell histiocytosis, inflammatory myofibroblastic tumor, or desmoid/dermoid/epidermoid tumors.
Histopathology remains the gold standard for diagnosis, but due to the rarity of the disease, there are no standardized treatment protocols. Surgery is performed to prevent complications or improve prognosis, with a 10% rate of recurrence. Chemotherapy may be used in cases of involvement of internal organs, unsuccessful surgery, or in unresectable lesions.
Histopathology remains the gold standard for the diagnosis but due to the rarity of the disease there are no standardized treatment protocols. Surgery is performed to prevent complications or improve prognosis, with 10% of recurrence, and chemotherapy may be used in involvement of the internal organs, unsuccessful surgery or in unresectable lesions.
Conclusion
A 6-year-old girl with a right temporal mass was diagnosed with infantile myofibroma, a rare benign soft tissue tumor, after a biopsy was performed. The tumor was treated with surgery and, in some cases, chemotherapy may be used if the tumor involves internal organs, is not successfully removed with surgery, or is unresectable. Histopathology is the gold standard for diagnosis and there is a 10% recurrence rate after surgery.
References
- Fang-Ying K, Shun-Cheng H, Hock-Liew E, Jiin-Haur C, Wei-Jen C (2002) Solitary infantile myofibromatosis: Report of two cases. Chang Gung Med J 25: 393-398.
- https://www.orpha.net/consor/cgi bin/Disease_Search.php?lng=PT&data_id=3545&Disease_Disease_Search_diseaseGroup=Infantile-Myofibromatosis&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Infantile-myofibromatosis&title=Infantile-myofibromatosis&search=Disease_Search_Simple
- https://rarediseases.org/rare-diseases/infantile-myofibromatosis/?filter=ovr-ds-resources
Citation: Miranda SB, Nogueira MV, Maia I, Maia A (2023) A Solitary Infantile Myofibroma – Case Report. J Neonatol Clin Pediatr 10: 103.
Copyright: © 2023 Sofia Brandão Miranda, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.