A Variant In The HNF4A Gene Causing MODY-1 In A Teenage Girl: Optimal Glycaemic Control After Transition To Sulfonylurea
*Corresponding Author(s):
Gabriela ReisPediatrics Service, José Joaquim Fernandes Hospital, Baixo Alentejo Local Health Unit, Beja, Portugal
Email:gabrielarmesquita90@gmail.com
Abstract
Maturity-Onset Diabetes of the Young (MODY) is a group of monogenic autosomal dominant forms of diabetes, characterized by early-onset of hyperglycemia, and typically the presence of a family history of diabetes in more than one generation. These features are also present in Type 1 and Type 2 Diabetes Mellitus, which frequently leads to misdiagnosis. There are various MODY subtypes, caused by mutations in 14 known genes, which share a primary defect in insulin secretion.
We describe the case of a teenage girl diagnosed with diabetes at 11-years-old whose father had a personal history of diabetes diagnosed at age 10. Therapy with insulin and metformin was started, with no improvement in metabolic control. She maintained poor metabolic control in the subsequent years, with records of therapeutic noncompliance. Following genetic testing, the patient’s diabetes was reclassified as MODY-1, and the treatment was changed from insulin to sulfonylurea with good response.
In recent decades, there has been a great Progress in the area of molecular genetics, which has led to the production of high-capacity and lower-cost techniques, allowing for a more accurate classification of patients and their families, and a more personalized approach to treatment.
Keywords
MODY-1; HNF4A; Diabetes; Mutation; Sulfonylurea
Introduction
Maturity-Onset Diabetes of the Young (MODY) is the most frequent type of monogenic diabetes, followed by neonatal or syndromic diabetes [1-2]. It is considered a group of endocrine disorders with clinical and genetic heterogeneity, caused by mutations that affect a sole gene involved in the pancreatic beta cell function [3]. Currently, MODY accounts for 1-2% of all cases of diabetes in Europe, with an estimated prevalence of 21 to 45/1,000,000 children [4-5].
Advances in the area of molecular genetics have led to the identification of 14 subtypes of MODY [6-8]. The most frequent subtypes are caused by mutations in genes encoding glucokinase (GCK-MODY-2), hepatocyte nuclear factor-1 alpha (HNF1A-MODY-3) or hepatocyte nuclear factor-4 alpha (HNF4A-MODY-1) [9-10].
MODY is characterized by early-onset (typically earlier than 25 years) of noninsulin-dependent diabetes and autosomal dominant inheritance (family history of diabetes in two or more successive generations) [1, 11]. However, these central clinical features may overlap with the ones present in Type 1 Diabetes Mellitus (T1DM) or Type 2 Diabetes Mellitus (T2DM), and consequently lead to misclassification of a significant percentage of MODY individuals, particularly at the beginning of the disease [11, 12].
It has been established that about 5% of patients diagnosed with diabetes before 45-years-old have MODY [12]. A pediatric study reported that 36% and 51% of individuals with MODY had been misdiagnosed with T1DM and T2DM, respectively [5].
A correct recognition of MODY is very important, since pharmacotherapy depends on the MODY subtype.
The HNF4A gene is a transcription factor involved in glucose metabolism, that is expressed predominantly in the liver, but in pancreatic islets cells and kidney as well [13]. HNF4A mutations are the cause of MODY-1 and constitute about 10% of MODY cases and currently over 103 mutations have been described [14].
MODY-1 is characterized by fetal macrosomia, transitory neonatal hyperinsulinemic hypoglycemia, gradual development of hyperglycemia, and onset of diabetes mellitus in adolescence or early adulthood. Adult patients with MODY-1 are better treated with oral sulfonylureas [1, 12]. In the pediatric population, there is limited evidence regarding sulfonylurea therapy.
Patients should be informed about the risk of micro and macrovascular complications [1].
The authors present the clinical course and pharmacological treatment of a case of MODY-1 following the genetic diagnosis, and discuss the importance of a correct classification of diabetes based on the exact molecular defect.
Case Report
A previously healthy 11-years-old girl presented with a one-month history of polyuria, polydipsia, nocturia and recurrent episodes of transitory abdominal pain. There were no complaints of fatigue or weight loss.
She was the first child of non-consanguineous parents, born at full term by vaginal delivery with a birth weight of 3760g (90th percentile). Since 5-years-old her weight was between 85th-97th percentiles.
Her 43-year-old father was diagnosed with T1DM at 10-years-old and was recently diagnosed with diabetic retinopathy. His diabetes was controlled with oral hypoglycemic agents (OHA), such as metformin and glibenclamide until 23-years-old, and later with insulin (determir and glulisine). Furthermore, her paternal grandmother also had diabetes from a young age, and was being treated with insulin and OHA (Figure 1).
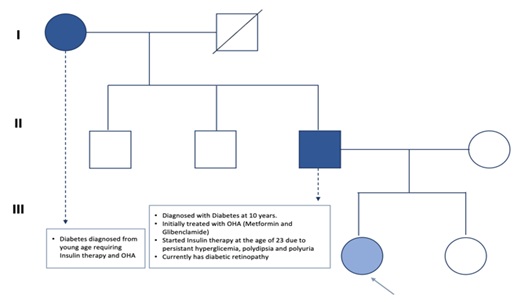 Figure 1: Our patient’s family genogram. In the first generation (I), her grandmother, diagnosed with Diabetes, has a negative genetic study. (II) In the second, her father, diagnosed with diabetes since 10 years, has a positive genetic study with the same variant. (III) Her mother has a negative genetic study. Our patient is marked with an arrow.
Figure 1: Our patient’s family genogram. In the first generation (I), her grandmother, diagnosed with Diabetes, has a negative genetic study. (II) In the second, her father, diagnosed with diabetes since 10 years, has a positive genetic study with the same variant. (III) Her mother has a negative genetic study. Our patient is marked with an arrow.
Due to the suspicion of MODY she started insulin glargine and metformin 500mg bid. Four months later, she required multiple daily insulin administrations. Auto-immunity study was negative for glutamate decarboxylase autoantibodies, insulin autoantibodies, zinc transporter autoantibodies and islet of Langerhans autoantibodies. The genetic study of this patient has been previously reported [10] and revealed a heterozygous variant (c.602A>C; p(His201Pro)) in the HNF4A gene. Subsequently, a genetic study was also performed on the father, the mother and the grandmother, and the same variant was found only in the father.
She maintained poor metabolic control (HbA1c 9.8%-11.2%) in the two subsequent years, with records of poor therapeutic compliance.
At 14-years-old, she was on metformin 1500 mg/day and intensive insulin regimen (glargine 38 units and rapid analogue to meals; total daily dose of 1.2 units/kg), with HbA1c of 8.6%. At this phase, a therapeutic transition regimen was initiated with close monitoring of capillary blood glucose: insulin glargine was replaced by insulin degludec and gliclazide 20 mg/day was started. Two weeks later, given the stable and on-target glycemic profile (70-180 mg/dL), the rapid-acting insulin was discontinued and gliclazide dose was adjusted to 40 mg/day. Progressive adjustments were made, and there was an improvement in therapeutic adherence. Six months later, she is under metformin 2000 mg, insulin degludec 30 units (total daily dose of 0.4 units/kg) and gliclazide 120 mg/day. She has an HbA1c of 6.9% and on flash glucose, monitoring 72% of values are on target, 26% are above target and 2% are below target (Figure 2).
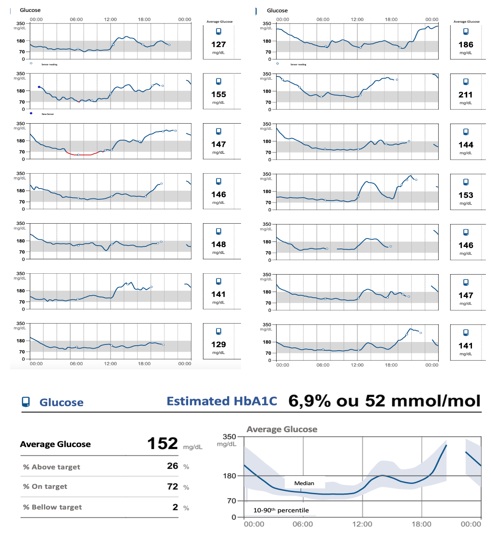 Figure 2: Patient’s glycaemic profile based on flash glucose monitoring, six months after starting gliclazide and replacing insulin glargine by insulin degludec.
Figure 2: Patient’s glycaemic profile based on flash glucose monitoring, six months after starting gliclazide and replacing insulin glargine by insulin degludec.
The authors followed the transition guide from the Royal Devon and Exeter National Health Service Foundation Trust, which is the United Kingdom´s reference center for genetic testing of monogenic diabetes [15, 16]. It should be noted that her father underwent an identical therapeutic transition regimen, also with metabolic improvement.
During the 3 years of follow-up, the patient never had evidence of micro and macrovascular complications. She maintains obesity (BMI 29.49 kg/m2) without clinical signs of insulin resistance. Blood insulin and C-peptide levels always remain in the normal range (Figure 3).
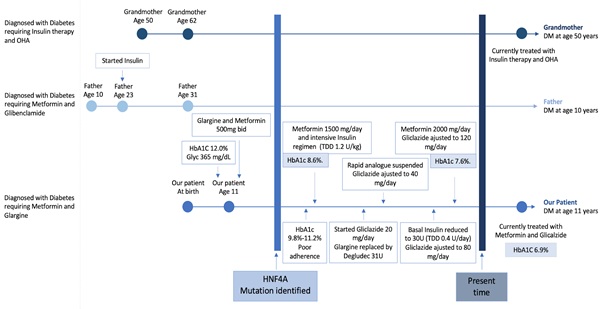 Figure 3: Timeline depicting the three phenotypes of the family. DM: Diabetes Mellitus; OHA: Oral Hipoglicemic Agents; TDD: Total Daily Dose; U: Units.
Figure 3: Timeline depicting the three phenotypes of the family. DM: Diabetes Mellitus; OHA: Oral Hipoglicemic Agents; TDD: Total Daily Dose; U: Units.
Discussion
Differential diagnosis between MODY, T1DM and T2DM is often difficult, as clinical features may overlap. Different from T1DM, MODY presents with persistent endogenous insulin secretion and frequently a noticeable family history of diabetes in a parental line [11]. Differently from T2DM, MODY does not have obvious obesity or evidence of insulin resistance [11, 17]. Therefore, in MODY patients, it is atypical the presence of diabetic ketoacidosis (DKA) [8], there is low necessity of exogenous insulin (
Correct differentiation of MODY from T1DM and T2DM is important to determine the treatment and prognosis of patients, and to detect family members at risk [18]. A study realized in the United Kingdom showed that patients had a delay of 13 years from being diagnosed with diabetes to receiving the correct diagnosis of MODY [4-6]. Moreover, it has been estimated that around 80% of individuals with MODY are misdiagnosed at presentation with T1DM or T2DM [4-6]. A recent study performed in the United States with children diagnosed with MODY by molecular methods described that, before the genetic diagnosis, 36% were treated for T1DM, 51% for T2DM, and only 24% for MODY [5, 6].
The diagnosis of MODY in a patient often results in genetic testing and diagnosis of other family members, which contribute to prompt treatment of their relatives [1]. A rapid advance in molecular genetics has led to the introduction of new high-capacity sequencing technologies and with it the hope of lowering the costs [7].
In a Portuguese multicentric study [10], which evaluated the genetic mutations in a group of patients with clinical suspicion of MODY, the authors identified 20 mutations, of which seven were novel mutations. These variants were not identified in healthy Portuguese controls. Most mutations in this study were missense, which is consistent with other European studies [14], and were predicted to be deleterious. Missense mutations may result in alterations of secondary structures, affecting protein stability or resulting in the loss of important catalytic domains.
There is current data on the success of transfer to sulfonylurea treatment in individuals with MODY-1 after genetic diagnosis [19, 20]. In those with MODY-1, shorter duration of disease, lower HbA1c and lower BMI at genetic diagnosis predicted successful treatment with sulfonylurea or diet alone. Sheperd et al. [19] suggest that, in individuals with MODY-1 with a longer duration of disease (more than 11 years) at time of genetic testing, rather than discontinuing current treatment, a sulfonylurea should be added to existing therapy, particularly in those with overweight/obesity and in those with higher HbA1c.
Progressive loss of pancreatic beta cell function is a hallmark of MODY-1, resulting in increased blood glucose and increased treatment requirements over time. The lack of standardized treatment guidelines for individuals who require additional second-line therapy may also lead to suboptimal glycemic control. Prompt transfer to sulfonylureas, allowing for optimal glycemic control soon after diagnosis of diabetes, may lower the risk of future adverse outcomes.
There are some particularities in this case that the authors would like to highlight, for example, the importance of therapeutic adherence, which in this case was initially a bias. Also, there are some aspects of the clinical presentation that are not so common among MODY patients, such as obesity. The diagnosis of this adolescent also allowed the reclassification of the father's diabetes, who had a twenty-year-history of insulin treatment and had already developed retinopathy.
In conclusion, the correct identification and classification of patients with MODY may have a remarkable clinical and social impact. It enables better therapeutic choices, more rigorous metabolic control, improved quality of life, adherence to therapy and screening of other affected family members.
Thus, the possibility of a diagnosis of MODY should be considered in any patient with an unusual course of diabetes that does not resemble T1DM or T2DM, even years after the onset of diabetes [11].
This case report also highlighted the importance of advances in genetic testing in diabetes, which provide individualized treatment based on an accurate diagnosis [6]. It is important to identify and describe novel mutations, for a better knowledge of the pathophysiology and clinical presentation of new forms of monogenetic diabetes and for a more personalized approach to the treatment, follow-up, and genetic counselling of patients [10].
Disclosure
The authors have no conflict of interests or financial disclosures.
References
- Fajans SS, Bell GI (2011) MODY: history, genetics, pathophysiology, and clinical decision making. Diabetes Care. 34: 1878-84.
- Aarthy R, Aston-Mourney K, Mikocka-Walus A, Radha V, Amutha A, et al. (2021) Clinical features, complications and treatment of rarer forms of maturity-onset diabetes of the young (MODY) - A review. J Diabetes Complications. 35: 107640.
- Sanyoura M, Philipson LH, Naylor R (2018) Monogenic Diabetes in Children and Adolescents: Recognition and Treatment Options. Curr Diab Rep. 18: 58.
- Shields BM, Hicks S, Shepherd MH, Colclough K, Hattersley AT, et al. (2010) Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia. 53: 2504-8.
- Pihoker C, Gilliam LK, Ellard S, Dabelea D, Davis C, et al. (2013) Prevalence, characteristics and clinical diagnosis of maturity onset diabetes of the young due to mutations in HNF1A, HNF4A, and glucokinase: results from the SEARCH for Diabetes in Youth. J Clin Endocrinol Metab. 98: 4055-62.
- Anik A, Çatli G, Abaci A, Böber E (2015) Maturity-onset diabetes of the young (MODY): an update. J Pediatr Endocrinol Metab. 28: 251-63.
- Urakami T (2019) Maturity-onset diabetes of the young (MODY): current perspectives on diagnosis and treatment. Diabetes Metab Syndr Obes. 12: 1047-1056.
- Oliveira SC, Neves JS, Pérez A, Carvalho D (2020) Maturity-onset diabetes of the young: From a molecular basis perspective toward the clinical phenotype and proper management. Endocrinol Diabetes Nutr (Engl Ed). 67: 137-147.
- Baldacchino I, Pace NP, Vassallo J (2020) Screening for monogenic diabetes in primary care. Prim Care Diabetes. 14: 1-11.
- Alvelos MI, Gonçalves CI, Coutinho E, Almeida JT, Bastos M, et al. (2020) Maturity-Onset Diabetes of the Young (MODY) in Portugal: Novel GCK, HNFA1 and HNFA4 Mutations. J Clin Med. 9: 288.
- Urbanova J, Brunerova L, Broz J (2019) How can maturity-onset diabetes of the young be identified among more common diabetes subtypes?. Wien Klin Wochenschr 131: 435-441.
- Thanabalasingham G, Pal A, Selwood MP, Dudley C, Fisher K, et al. (2012) Systematic assessment of etiology in adults with a clinical diagnosis of young-onset type 2 diabetes is a successful strategy for identifying maturity-onset diabetes of the young. Diabetes Care. 35: 1206-12.
- Stoffel M, Duncan SA (1997) The maturity-onset diabetes of the young (MODY1) transcription factor HNF4alpha regulates expression of genes required for glucose transport and metabolism. Proc Natl Acad Sci U S A. 94:13209-14.
- Colclough K, Bellanne-Chantelot C, Saint-Martin C, Flanagan SE, Ellard S (2013) Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha and 4 alpha in maturity-onset diabetes of the young and hyperinsulinemic hypoglycemia. Hum Mutat. 34: 669-85.
- Pearson ER, Flechtner I, Njølstad PR, Malecki MT, Flanagan SE, et al. (2006) Neonatal Diabetes International Collaborative Group. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med. 355: 467-77.
- Guidance for transferring HNF1A MODY or HNF4A MODY patients from insulin to sulphonylureas (2017) ROYAL DEVON & EXETER NHS FOUNDATION TRUST; Available in online.
- Nkonge KM, Nkonge DK, Nkonge TN (2020) The epidemiology, molecular pathogenesis, diagnosis, and treatment of maturity-onset diabetes of the young (MODY). Clin Diabetes Endocrinol. 6: 20.
- Schober E, Rami B, Grabert M, Thon A, Kapellen T, et al. (2009) Phenotypical aspects of maturity-onset diabetes of the young (MODY diabetes) in comparison with Type 2 diabetes mellitus (T2DM) in children and adolescents: experience from a large multicentre database. Diabet Med. 26: 466-73.
- Shepherd MH, Shields BM, Hudson M, Pearson ER, Hyde C, et al. (2018) A UK nationwide prospective study of treatment change in MODY: genetic subtype and clinical characteristics predict optimal glycaemic control after discontinuing insulin and metformin. Diabetologia. 61: 2520-2527.
- Chandran S, Rajadurai VS, Hoi WH, Flanagan SE, Hussain K, et al. (2020) A Novel HNF4A Mutation Causing Three Phenotypic Forms of Glucose Dysregulation in a Family. Front Pediatr. 8: 320.
Citation: Reis G, Martins S, Antunes A, Lemos MC, Gomes MM (2023) A Variant In The HNF4A Gene Causing MODY-1 In A Teenage Girl: Optimal Glycaemic Control After Transition To Sulfonylurea. J Clin Stud Med Case Rep 10: 0154.
Copyright: © 2023 Gabriela Reis, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.