Adult-Onset Metastatic Pancreatic Ganglioneuroblastoma with ALK Gain-of-Function Mutation P.R1275Q Treated with Lorlatinib
*Corresponding Author(s):
Cingarlini SaraSection Of Oncology, Department Of Medicine, University Of Verona School Of Medicine And Verona University Hospital Trust, Verona, Italy
Email:sara.cingarlini@aovr.veneto.it
Introduction
Ganglioneuroblastoma (GNB) is a very rare tumor entity that typically occurs in pediatric patients, with most cases occurring in children aged up to four years. [1] GNBs are composite tumor, embryologically derived from neural crest cells, containing both primitive neuroblasts and mature ganglion cells; they are considered to have an intermediate metastatic potential between the more aggressive neuroblastomas and the benign ganglioneuroma. [2] They can develop in any organs of the sympathetic nervous system. According to few case reports and small case series reported in literature, the majority of sympathetic paragangliomas arise below the diaphragm in retroperitoneum or adrenal glands; less frequently GNB originate in the thorax or in the head and neck district. [3] Here, we present the clinical course and multimodal therapeutically strategies in an adult-onset pancreatic ganglioneuroblastoma.
Case Presentation
This is the unique case of an asymptomatic 43-year-old female patient diagnosed with pancreatic ganglioneuroblastoma (GNB) in November 2015, when she underwent a routine abdominal CT scan with incidental finding of a voluminous cephalopancreatic mass. The patient was referred to our Institution’s Oncology Department for further management. Diagnostic workup included an endoscopic ultrasonography (EUS) which described a 53 mm x 58 mm hypoechoic cephalopancreatic tumor with irregular margins and multiple internal calcifications; slightly enlarged loco regional lymph nodes were also reported. Cytological examination was consistent with the diagnosis of synaptophysin-positive neuroendocrine tumor, with 15-20% Ki-67 labeling index. Dual-tracer PET-CT scan with 68-Ga-DOTATOC and 18F-FDG showed moderate tumor SSTR expression (SUV max 14) and low hyperactive metabolism (SUV max 6), respectively. After multidisciplinary assessment, the pancreatic lesion was deemed resectable; therefore, in January 2016 the patient underwent a duodenocephalopancreatectomy with pylorus preservation (DCP-PP). The histopathological report described a well circumscribed, multi-nodular, with a calcific inner core solid tumor, 50 mm in its greater dimension. Histopathologic findings were consistent with peripheral neuroblastic tumor with features of nodular ganglioneuroblastoma. (Figure 1) No lymph node metastasis was reported. Surgical resections were R0. Vascular invasion was present. No secondary lesion was detected in the post-operative CT-scan; therefore, given the lack of evidence proving benefit for adjuvant treatment in this setting, no further action was taken. In January 2019, three years after surgery, MRI of the abdomen detected a single interaortocaval lymph node metastasis; a 68-Ga-DOTATOC PET/TC confirmed a moderate uptake (SUV max 7). A loco regional approach with stereotactic radiotherapy was chosen to treat this single lymph node metastasis (dose 50 Gy in 10 fractions). In August 2019, MRI detected a 10-mm hepatic lesion in VII liver segment. This single lesion was successfully treated with radiofrequency thermal ablation (RFTA), with good disease control. However, in February 2021 the patient complained progressively worsening low back pain. An MRI documented multiple bone metastases, the largest in the left proximal femur (3 cm in maximal length), and other lesions in sacrum, lumbar spine and bilateral iliac bones. Bone lesions had a moderate tracer uptake at a 68-Ga-DOTATOC PET/CT, and mild hyper metabolism at (18) F-FDG PET/CT (SUV max 5.7). Given the symptomatic osseous disease progression and the positivity of 68-Ga-DOTATOC uptake by bone metastasis, the patient was evaluated for peptide receptor radionuclide therapy (PRRT). Four 177-Lu-PRRT cycles were administered from March to October 2021; during the radionuclide therapy, the patient reported a significant clinical benefit in term of decreased bone pain. However, shortly after completion of the treatment she complained rapidly increasing diffuse pain, indeed post-treatment SPECT/CT showed diffuse bone progression of disease, later on confirmed by a spine MRI and abdominal-thoracic CT scan.
 Figure 1: Hematoxylin-eosin histopathological images. (A) Coexistence of neuroblastic and ganglion cells 20x other filed, (B) Details on vascular invasion 20x, (C) Ganglion cells 20x and (D) Neuroblastic cells 20x.
Figure 1: Hematoxylin-eosin histopathological images. (A) Coexistence of neuroblastic and ganglion cells 20x other filed, (B) Details on vascular invasion 20x, (C) Ganglion cells 20x and (D) Neuroblastic cells 20x.
At this point a Next Generation Sequencing (NGS) analysis on the tumoral tissue obtained from the DCP-PP surgery was performed. The analysis by NGS was performed by using an in-house multigene next generation sequencing panel. Tumoral tissue obtained by surgical specimen was fixed in formalin-fixed and paraffin-embedded tissue, and DNA extraction was performed.
A gain-of-function mutation involving the ALK gene with an allelic frequency of 49% was identified: p.R1275Q (Substitution - Missense, position 1275, (R→Q). In addition, a variant of unknown significance (VUS) mutation in EPHA3 gene was found (p.P845H). The tumor mutation burden (TMB) was 5.56 Mut/Mb.
Table 1 The ALK gene gain-of-function mutation p.R1275Q is known to be one of the most common oncogenic mutations in ALK-altered GNB [4].
|
TMB |
MSI |
SNV |
VUS |
CNV |
Structural Variations |
|
5,56 |
MSS |
ALK |
EPHA3 |
ATRX |
LOH whole chr 3, 10, 12, 14, 15, 16 |
|
p.R1275Q |
p.P845H |
large home del |
LOH9P, 11q22.3-25, Xq13.1-q28 |
||
|
freq 45 |
freq 42 |
exon 2-9 |
|||
|
class 5 |
class 3 |
class 5 |
|||
|
TMB= Tumor mutational burden, SNV= Small nucleotide variations, VUS= Variants of uncertain significance, CNV= Copy number variations. |
|||||
|
<> Refers to ACMG/AMP classification. |
|||||
Table 1: NGS Report.
Therefore, given the exceptional rarity of the disease and the result of the NGS testing, we discussed this clinical case at the Molecular Tumor Board (MTB) of our Institution.
Our patient’s case was submitted to the local molecular tumor board of our Institution and, after a detailed discussion, Lorlatinib was chosen as the best option, given the high burden of symptomatic disease. Therefore, the patient started treatment with Lorlatinib 100mg/day in December 2021. After few months of treatment, in April 2022, a 68-Ga-DOTATOC PET/CT showed a stunning response of disease with respect to the previous one performed in February 2022; this was fully consistent with the clinical benefit reported by the patient. The patient reports complete regression of bone pain. In December 2022, restaging assessed radiologic maintained partial response and complete disappearance of pathologic gallium uptake. (Figures 2-3) As of January 2023, treatment is still ongoing. (Figure 4).
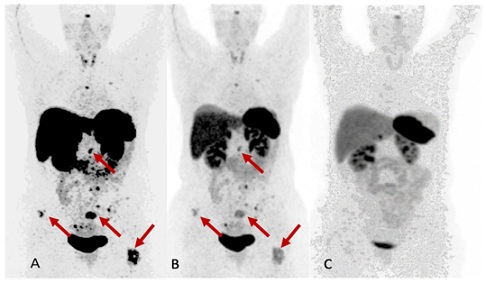 Figure 2: Panoramic view of the Gallium PET-TAC during treatment. Picture (A) at baseline, (B) At Feb 22 and (C) at Dec 22.
Figure 2: Panoramic view of the Gallium PET-TAC during treatment. Picture (A) at baseline, (B) At Feb 22 and (C) at Dec 22.
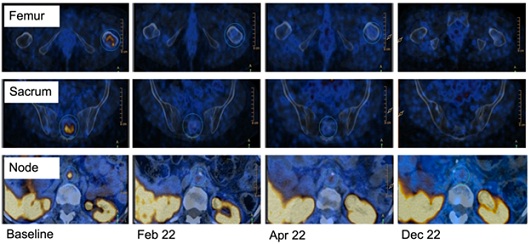 Figure 3: Details of the different disease sites at Gallium PET imaging during treatment.
Figure 3: Details of the different disease sites at Gallium PET imaging during treatment.
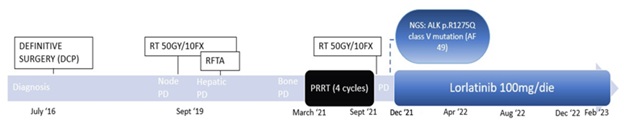 Figure 4: Timeline of patient’s clinical history.
Figure 4: Timeline of patient’s clinical history.
Discussion
Despite being quite common among children, adult-onset ganglioneuroblastoma is a very rare tumor that may arise in any location along the sympathetic nervous system, with less than 50 cases reported in the literature.1 The majority of reported adult-onset GNBs are described in case reports or small case series, highlighting the rarity of this neoplasm. It is recognized that survival of stage IV ganglioneuroblastoma patients is poor. In the English literature, only one patient with stage IV GNB survived up to 5 years after diagnosis. [5] When a GNB is found in an adult, it usually is classified as high-risk tumor and is associated with a poor prognosis. [6] The currently recognized negative prognostic factors in GNB are older age at diagnosis, advanced stage, retroperitoneal primary tumor, N-MYC amplification and DNA ploidy [5, 7].
Given the rarity of adult-onset ganglioneuroblastoma, treatment recommendations usually follow pediatric guidelines. In pediatric populations, patients with advanced disease have responded to first-line multi-drug chemotherapy with cyclophosphamide, vincristine and doxorubicin, or combinations with platinum and etoposide. Classically accepted treatments consist of any combination of surgery, radiotherapy, chemotherapy. [1] However pediatric treatments result often too toxic in adult patients.
In recent years, metabolic radiotherapy has been used in the treatment of advanced neuroblastomas by exploiting the ability of somatostatin receptor-positive tumours to fix somatostatin analogue peptides coupled with a radionuclide, most frequently 177-Lu. [5, 7] PRRT has been performed for unresectable neurobastomas in small series of pediatric patients and in phase I clinical trials, with encouraging results [8].
Thanks to the diffusion of Next Generation Sequencing (NGS), detection of gene mutations and identification of patients that will benefit from a particular drug have significantly improved in the last decade. Tyrosine kinase inhibitors (TKIs) have been developed to target specific gene alterations associated with tumor proliferation. The ALK (anaplastic lymphoma kinase) gene, which encodes a receptor-type protein tyrosine kinase, is frequently altered in neuroblastic tumors, accounting for 8% of total neuroblastic tumor’s population and up to 14% of high-risk population [9]. The two most common mutations are ALK R1275Q and ALK F1174L, which result in constitutive active forms of this protein. These observations highlight the role of ALK as a driver oncogene in both primary and relapsed neuroblastic tumors. [10, 11] Although no specific, mutation showed different prognostic significance over the others, [12] they display a different sensitivity to ALK inhibitors. ALK-mutant GNB display different sensitivity to ALK-inhibitors according to the mutation they carry, and the magnitude of benefit to be expected by target treatments differs widely depending on the mutation and the ALKi. [9] Lorlatinib, a third-generation inhibitor of ALK and ROS1, exhibits improved efficacy over first-generation crizotinib, towards three of the most common ALK mutations in neuroblastic tumors, which include R1275Q, as shown by in vitro kinase activity. [13] It was also demonstrated that brigatinib abrogates ALK activation in neuroblastoma cell lines in vitro biochemical assays. [14] Moreover, exceptional preclinical activity with third-generation ALKi Lorlatinib was reported and clinical trials are ongoing [15].
The exact role of ALK inhibitors in neuroblastic tumors will be determined in the next few years, although it is expected that long-term use of TKI will require a combination of target agents acting also on downstream pathways, to overcome secondary resistance mechanisms to ALK inhibitors [10].
Conclusion
Our case illustrates a multimodality therapeutic approach to an advanced cephalopancreatic ganglioneuroblastoma, relapsed three years after radical surgery and subsequently treated with a multimodality-personalized approach, including stereotactic radiotherapy, radiofrequency thermal ablation, and PRRT and target therapy. By incorporating new technologies into cancer diagnostic, patients with otherwise limited therapy options could be offered target therapies, resulting in possible improvement in disease control and survival rates and acceptable quality of life. To the best of our knowledge, no previously reported cases in the literature have described a metastatic, ALK-mutated, adult-onset pancreatic ganglioneuroblastoma.
Moreover, this case highlights the importance of undertaking a patient-tailored therapeutic approach in patients with rare tumors.
References
- Lonie J, Boles R, Boldery J (2019) Adrenal ganglioneuroblastoma in an adult. ANZ Journal of Surgery. 89: 129-130.
- Mehta N, Tripathi RP, Popli MB, Nijhawan VS (1997) Bilateral intraabdominal ganglioneuroblastoma in an adult. British Journal of Radiology. 70: 96-98.
- Lam AKY (2017) Update on Adrenal Tumours in 2017 World Health Organization (WHO) of Endocrine Tumours. Endocrine Pathology. 28: 213-227.
- Mossé YP, Laudenslager M, Longo L, Cole KA, Wood A, et al. (2008) Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 455: 930-935.
- Risum S, Knigge U, Langer SW (2017) Hitherto unseen survival in an ALK-positive-patient with advanced stage adult ganglioneuroblastoma treated with personalized medicine. Clinical Case Reports. 5: 2085-2087.
- Jrebi NY, Iqbal CW, Joliat GR, Sebo TJ, Farley DR (2014) Review of our experience with neuroblastoma and ganglioneuroblastoma in adults. World J Surg. 38: 2871-2874.
- Mousa AM, Shokouh-Amiri MH, Shah LM, Garzon S, Xie KL (2020) Adult-onset ganglioneuroblastoma of the posterior mediastinum with osseous metastasis. Radiology Case Reports. 15: 1676-1682.
- Watanabe N, Nakanishi Y, Kinukawa N, Ohni S, Obana Y, et al. (2014) Expressions of somatostatin receptor subtypes (SSTR-1, 2, 3, 4 and 5) in neuroblastic tumors; special reference to clinicopathological correlations with International Neuroblastoma Pathology Classification and outcomes. Acta Histochem Cytochem. 47: 219-229.
- Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, et al. (2014) ALK Mutations Confer Differential Oncogenic Activation and Sensitivity to ALK Inhibition Therapy in Neuroblastoma. Cancer Cell. 26: 682-694.
- Trigg RM, Turner SD (2018) ALK in neuroblastoma: Biological and therapeutic implications. Cancers (Basel). 10: 113.
- Ueda T, Nakata Y, Yamasaki N, Oda H, Sentani K, et al. (2016) ALK R1275Q perturbs extracellular matrix, enhances cell invasion and leads to the development of neuroblastoma in cooperation with MYCN. Oncogene. 35: 4447-4458.
- O’Donohue T, Gulati N, Mauguen A, Kushner BH, Shukla N, et al. (2021) Differential Impact of ALK Mutations in Neuroblastoma . JCO Precision Oncology. 5: 492-500.
- Infarinato NR, Park JH, Krytska K, Ryles HT, Sano R, et al. (2016) The ALK/ROS1 inhibitor PF-06463922 overcomes primary resistance to crizotinib in ALK-driven neuroblastoma. Cancer Discovery. 6: 96-107.
- Siaw JT, Wan H, Pfeifer K, Rivera VM, Guan J, et al. (2016) Brigatinib, an anaplastic lymphoma kinase inhibitor, abrogates activity and growth in ALK-positive neuroblastoma cells, Drosophila and mice. Oncotarget. 7: 29011-29022.
- Tucker ER, Poon E, Chesler L (2019) Targeting MYCN and ALK in resistant and relapsing neuroblastoma. Cancer Drug Resistance. 2: 803-812.
Citation: Alice R, Michele B, Claudio L, Andrea M, Sara C (2023) Adult-Onset Metastatic Pancreatic Ganglioneuroblastoma with ALK Gain-of-Function Mutation P.R1275Q Treated with Lorlatinib. J Clin Stud Med Case Rep 10: 0150.
Copyright: © 2023 Rossi Alice, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.