Journal of Non Invasive Vascular Investigation Category: Clinical
Type: Research Article
AMP-Activated Protein Kinase Inhibits Arterial Smooth Muscle Cell Proliferation in Vasodilator-Stimulated Phosphoprotein-Dependent Manner
*Corresponding Author(s):
David A TulisDepartment Of Physiology, Brody School Of Medicine, East Carolina University, Greenville, NC, 27834, United States
Tel:+1 2527442771,
Email:tulisd@ecu.edu
Received Date: Jun 23, 2016
Accepted Date: Aug 04, 2016
Published Date: Aug 23, 2016
Abstract
Abnormal arterial smooth muscle (ASM) growth is a major contributor to the etiology of many occlusive vascular disorders, yet to date no target or process capable of completely controlling pathologic ASM growth has been identified. In turn, lack of precision in identifying full causation of abnormal ASM growth has limited utility of feasible biomarkers for early identification and perhaps prophylaxis for ensuing cardiovascular events. In this light we recently reported capacity of the metabolic sensor AMP-activated protein kinase (AMPK) to inhibit ASM cell proliferation and migration under in vitro and in vivoconditions. Microfilament-associated vasodilator-stimulated phosphoprotein (VASP) has also been implicated in the control of pathologic ASM cell growth via dynamic interaction with the actin cytoskeleton and focal adhesion complexes, and new findings published by our lab suggest interdependence between AMPK and VASP in ASM. In the current study we hypothesized that AMPK reduces ASM cell growth through mechanisms dependent upon VASP and theorized that VASP phosphorylation status may represent a useful biological marker for AMPK-related metabolic and cardiovascular disorders. In rat A7r5 ASM cells, activation of AMPK by the AMP-mimetic AICAR significantly increased phosphorylation of VASPThr278 and the protein kinase A target VASPSer157, proposed VASP inhibitory sites [1,2], with little effect on the protein kinase G marker VASPSer239 after 60 minutes. At this time point AICAR-stimulated AMPK doubled G-actin to F-actin ratio (G:F), significantly reduced (~40%) catalytic Tyr397 phosphorylation of focal adhesion kinase (FAK) and significantly increased (~100%) cytostatic paxillin compared to vehicle controls. Functionally, AMPK significantly inhibited PDGF-stimulated transwell migration of ASM cells after 18 hours and serum-stimulated cell cycle progression after 24 hours compared to vehicle controls. To determine VASP-dependency of these events we performed studies using Lentiviral non-targeting control (Lv-NTC) and short hairpin RNA (Lv-shRNA)-mediated VASP-deficient ((-)VASP) ASM cells. Results showed that (-)VASP cells displayed significantly reduced (~75%) VASP expression (normalized to DNA content) 48 hours post-infection compared to Lv-NTC cells and significantly diminished AMPK-stimulated VASPSer157 and VASPThr278compared to controls. (-)VASP cells demonstrated significantly increased G:F actin ratios yet had similar levels on FAKTyr397 and paxillin compared to AMPK-stimulated cells. Interestingly, VASP deficiency failed to markedly alter the inhibitory effects of AMPK on ASM cell migration yet fully reversed AMPK-mediated cell cycle inhibition compared to controls. These findings suggest that AMPK has ability to reduce ASM cell growth by inhibiting VASP-directed actin cytoskeletal dynamics and provide rationale for continued exploration of AMPK and VASP as targets for vascular growth control. Lastly, these new observations lend credence for VASP as a potential biological marker for AMPK-driven metabolic and associated cardiovascular disorders.
INTRODUCTION
Many disease processes including tumor angiogenesis, evolution of atherosclerotic plaques, transplant vasculopathy and restenosis after angioplasty or bypass grafting involve abnormal growth of blood vessels and in particular, arterial smooth muscle (ASM). According to the American Heart Association, cardiovascular disease (CVD) remains the number one cause of death in the United States and over half of these deaths involve pathologic ASM growth [1]. Despite wide-ranging efforts aimed at identifying and characterizing mechanisms involved in this ASM growth, our understanding of this process is deficient and continues to present a critical barrier to our understanding of key elements of vascular pathobiology and for the development of improved and targeted therapeutics to effectively combat occlusive CVD. Additionally, lack of identification and characterization of key elements behind CVD pathogenesis limits effectiveness and utility of biological markers for early detection of and, perhaps, prophylaxis against ensuing CVD-related complications.
Adenosine monophosphate-activated protein kinase (AMPK) is a ubiquitously expressed heterotrimeric Serine (Ser)/Threonine (Thr) kinase consisting of a catalytic α-subunit and two regulatory subunits β and γ [2,3]. AMPK responds to increased AMP: ATP under metabolic challenge as well as to chemical and physical stresses. Upon activation of AMPK, energy producing pathways are upregulated while energy consuming pathways are shut off or rendered minimally active [3,4]. Functionally, AMPK phosphorylates and inhibits many key metabolic enzymes such as glycogen synthase, HMG-CoA reductase, and acetyl CoA carboxylase [4,5], and AMPK has been implicated in the control of cardiac and vascular oxidative burden, fibrosis and growth [2,6-8]. In complement, we recently showed that AMPK has capacity to communicate with the cyclic AMP/protein kinase A (PKA) pathway in growth suppression of ASM under both in vitroand in vivo conditions [9-13].
Many elements control vessel wall biology and function including those that comprise the cellular architecture such as the extracellular matrix, cell-to-cell and focal adhesion contacts and the cytoskeleton. The actin cytoskeleton is a highly dynamic structure that plays critical roles in directing cellular signaling events and cellular functions. Actin-binding vasodilator-stimulated phosphoprotein (VASP) is a key cytoskeletal regulator that promotes actin polymerization by delivering monomeric globular (G) actin to the barbed end of growing F-actin [14-16]. VASP has been implicated in the control of intra- and extra-cellular signaling events associated with transcriptional activity as well as cell migration and proliferation [13-19]. Due to its role as a mediator of signal transduction, differential VASP phosphorylation has also been used as a readout of discrete protein kinase signaling [17,20-22], and we [9,10] and others [23] have documented capacity of AMPK to phosphorylate VASP at discrete Ser and/or Thr residues. It is therefore intriguing to consider VASP as a downstream target of Ser/Thr protein kinases including AMPK that is capable of inhibiting abnormal ASM growth and that may serve as a read out of AMPK-related cardiovascular disorders.
The purpose of this study was to test our hypothesis that AMPK inhibits ASM cell proliferation and migration through mechanisms dependent upon VASP. Using viral-driven VASP deficiency, novel findings show that AMPK suppresses ASM cell proliferation in VASP-specific manner. These new data also highlight VASP as a potential therapeutic target and biomarker to be used for detection and perhaps prevention of AMPK-related vascular growth disorders.
Adenosine monophosphate-activated protein kinase (AMPK) is a ubiquitously expressed heterotrimeric Serine (Ser)/Threonine (Thr) kinase consisting of a catalytic α-subunit and two regulatory subunits β and γ [2,3]. AMPK responds to increased AMP: ATP under metabolic challenge as well as to chemical and physical stresses. Upon activation of AMPK, energy producing pathways are upregulated while energy consuming pathways are shut off or rendered minimally active [3,4]. Functionally, AMPK phosphorylates and inhibits many key metabolic enzymes such as glycogen synthase, HMG-CoA reductase, and acetyl CoA carboxylase [4,5], and AMPK has been implicated in the control of cardiac and vascular oxidative burden, fibrosis and growth [2,6-8]. In complement, we recently showed that AMPK has capacity to communicate with the cyclic AMP/protein kinase A (PKA) pathway in growth suppression of ASM under both in vitroand in vivo conditions [9-13].
Many elements control vessel wall biology and function including those that comprise the cellular architecture such as the extracellular matrix, cell-to-cell and focal adhesion contacts and the cytoskeleton. The actin cytoskeleton is a highly dynamic structure that plays critical roles in directing cellular signaling events and cellular functions. Actin-binding vasodilator-stimulated phosphoprotein (VASP) is a key cytoskeletal regulator that promotes actin polymerization by delivering monomeric globular (G) actin to the barbed end of growing F-actin [14-16]. VASP has been implicated in the control of intra- and extra-cellular signaling events associated with transcriptional activity as well as cell migration and proliferation [13-19]. Due to its role as a mediator of signal transduction, differential VASP phosphorylation has also been used as a readout of discrete protein kinase signaling [17,20-22], and we [9,10] and others [23] have documented capacity of AMPK to phosphorylate VASP at discrete Ser and/or Thr residues. It is therefore intriguing to consider VASP as a downstream target of Ser/Thr protein kinases including AMPK that is capable of inhibiting abnormal ASM growth and that may serve as a read out of AMPK-related cardiovascular disorders.
The purpose of this study was to test our hypothesis that AMPK inhibits ASM cell proliferation and migration through mechanisms dependent upon VASP. Using viral-driven VASP deficiency, novel findings show that AMPK suppresses ASM cell proliferation in VASP-specific manner. These new data also highlight VASP as a potential therapeutic target and biomarker to be used for detection and perhaps prevention of AMPK-related vascular growth disorders.
MATERIALS AND METHODS
This investigation was approved by the East Carolina University Animal Care and Use Committee and conformed to the guide for the care and use of laboratory animals (US National Institutes of Health, Publication No. 85-23, revised 1996).
Materials
AICAR was purchased from Toronto Research Chemicals (North York, Ontario) and Invitrogen (Carlsbad, California). Scrambled, Lv-non-targeting controls (Lv-NTC) and Lv-shRNA (SMART vector 2.0) directed against full-length VASP were purchased from Dharmacon (Lafayette, CO). Primary antibodies targeted against VASP, VASPSer157,VASPSer239,VASPThr278, FAK, FAKTyr397, paxillin, α-tubulin and β-actin were purchased from Abcam (Cambridge, Massachusetts), ECM Biosciences (Versailles, Kentucky), or Cell Signaling (Danvers, Massachusetts). Antibodies were diluted 1:1000 for In-Cell and enhanced chemiluminescent (ECL) Western blotting, or 1:500 for immunofluorescent flow cytometry. Antibodies were diluted in IRDye blocking buffer (Rockland; Gilbertsville, Pennsylvania) for In-Cell Western blotting or 1% bovine serum albumin (BSA) in PBS (Gemini; Stoneham, MA) for immunofluorescence. IRDye secondary antibodies (1:1000; Rockland) and FITC- or TxRed-conjugated secondary antibodies (1:10,000; Rockland) were used for protein detection while Draq 5 (1:10000; Cell Signaling) and Sapphire 700 (1:1000; Li-Cor; Lincoln, Nebraska) were used for DNA staining and protein normalization. Propidium iodide (PI) and RNAse for cell cycle analysis were purchased from Invitrogen. Alexa Fluor-conjugated deoxyribonuclease I and phalloidin stains for G-actin and F-actin, respectively, were purchased from Invitrogen.
Methods
Cell culture: Rat A7r5 ASM cells (ATCC, Manassas, VA) were cultured under sterile conditions in Dulbecco’s Modified Eagles Medium (DMEM) supplemented with fetal bovine serum (FBS, 0.2-10%), 2 mM L-glutamine, a 1:500 dilution of 50 µg/mL Primocin (Invivogen) at 37ºC in 95% air/5% CO2 and were serially split and used through passage 6 as described [11,13,22].
Expression analyses: Cells were seeded in 96-well plates until confluent and treated with vehicle (DMSO) or AICAR (1 mM in DMSO), and after specified times total and phosphorylated protein expression was determined by ECL-based Western blotting on cell homogenates and by In-Cell Western blotting on intact, adherent cells as we have described [9-11,13,20-22]. In brief, fixed cells were permeabilized with 0.1% Triton-X, blocked with IRDye blocking buffer and treated with rabbit anti-rat primary antibodies for 1 hour at room temperature (RT). Target proteins were IR-labeled and DNA was stained for protein normalization. Fluorescence was detected and analyzed using Li-Cor Odyssey Infrared Imaging System and software. Additionally, ECL Western blots were performed on cell lysates to verify In-Cell Western data for select experiments as previously performed [9-11,13,20-22].
Immunofluorescence: Confluent ASM cells were treated with select pharmacologic agents for 1-24 hours and then trypsinized, fixed with 4% formalin, washed with PBS, permeabilized with 0.1% Triton-X in PBS, washed and blocked with 1% BSA in PBS. Select proteins were detected by primary/secondary conjugation in 1% BSA and analyzed by flow cytometry (C6 Flow Cytometer, Accuri) using CFlow Plus software (Accuri) [10,13]. Fluorescently-tagged phalloidin and deoxyribonuclease I were used to detect F-actin and G-actin, respectively, and were analyzed by flow cytometry [13].
Cell migration analyses: Following protocols previously described with minor modifications [13,20,21], ASM cells were seeded at 180,000 cells/ml in the upper chamber of a FluoroBlok transwell system (BD) in complete media and allowed to adhere. Cells were treated for 1 hour followed by staining with Cell Tracker Green (10 µM; Invitrogen). Serum-free media was applied to the upper chamber containing the same original treatment and 10 ng PDGF-β was applied to the bottom chamber as a chemoattractant [24]. Cell migration was assessed from time 0 through 18 hours by bottom-read fluorescence at 525 nm (Tecan Infinite M200) with each time point Relative Fluorescent Units (RFU) normalized to time=0 RFU for each respective condition. Net migration was calculated as a fold change of the total migration for each condition over total control (vehicle) migration at 18 hours.
Cell cycle analysis: Cells were plated in 12-well plates at 80,000 cells/well in complete media until ~50% confluent. Cells were quiesced in 0.5% FBS for 24 hours followed by treatment in complete growth media (DMEM, 10% FBS, Primocin) containing select pharmacologic agents for 24 hours. Cells were trypsinized, formalin-fixed and stained with PI (Invitrogen) per manufacturer’s recommendations [9,21]. The fraction of cells present in each phase of the cell cycle was assessed by flow cytometry.
Anti-VASP Lv-shRNA infection: Scrambled, NTC controls or Lv-shRNA (SMART vector 2.0) directed against the full-length VASP (Dharmacon Research, Lafayette, CO) were utilized to reduce expression of endogenous VASP. Infection efficiencies and appropriate multiplicity of infection (MOI) were determined per manufacturer’s guidelines (data not shown). Rat ASM cells were plated in 96-well plates at 40% confluence and switched to low serum (0.5%) following adherence. Cells were treated with Lv-shRNA SMART vector 2.0 directed against VASP (MOI 0.3-5), NTC or vehicle for 24 hours in antibiotic-free, low serum media. Media was changed to high glucose-DMEM, 15% FBS and penicillin/streptomycin mix after 24 hours, and on day 3 cells were expanded to 24-well plates and VASP mutants ((-)VASP cells) were selected by puromycin resistance for 24 hours. Stable cell lines were established and used for verification of VASP deficiency as well as for cellular signaling and migration and proliferation analyses.
Statistical analyses: Data were analyzed using Excel 2011/2013 (Microsoft) and Sigma Plot 11.2 (SPSS, Inc.). All data sets were tested for normal distribution and met the homogeneity prerequisites for Analysis of Variance (ANOVA). One-way ANOVA and Tukey’s post-hoc multiple comparison tests were used to detect changes between individual groups. Two-way ANOVA with multiple comparisons and Tukey’s post-hoc tests were used for migration and cell cycle analyses to detect significance between groups. Data are expressed as mean ± Standard Error of the Mean (SEM) with p<0.05 considered statistically significant.
Expression analyses: Cells were seeded in 96-well plates until confluent and treated with vehicle (DMSO) or AICAR (1 mM in DMSO), and after specified times total and phosphorylated protein expression was determined by ECL-based Western blotting on cell homogenates and by In-Cell Western blotting on intact, adherent cells as we have described [9-11,13,20-22]. In brief, fixed cells were permeabilized with 0.1% Triton-X, blocked with IRDye blocking buffer and treated with rabbit anti-rat primary antibodies for 1 hour at room temperature (RT). Target proteins were IR-labeled and DNA was stained for protein normalization. Fluorescence was detected and analyzed using Li-Cor Odyssey Infrared Imaging System and software. Additionally, ECL Western blots were performed on cell lysates to verify In-Cell Western data for select experiments as previously performed [9-11,13,20-22].
Immunofluorescence: Confluent ASM cells were treated with select pharmacologic agents for 1-24 hours and then trypsinized, fixed with 4% formalin, washed with PBS, permeabilized with 0.1% Triton-X in PBS, washed and blocked with 1% BSA in PBS. Select proteins were detected by primary/secondary conjugation in 1% BSA and analyzed by flow cytometry (C6 Flow Cytometer, Accuri) using CFlow Plus software (Accuri) [10,13]. Fluorescently-tagged phalloidin and deoxyribonuclease I were used to detect F-actin and G-actin, respectively, and were analyzed by flow cytometry [13].
Cell migration analyses: Following protocols previously described with minor modifications [13,20,21], ASM cells were seeded at 180,000 cells/ml in the upper chamber of a FluoroBlok transwell system (BD) in complete media and allowed to adhere. Cells were treated for 1 hour followed by staining with Cell Tracker Green (10 µM; Invitrogen). Serum-free media was applied to the upper chamber containing the same original treatment and 10 ng PDGF-β was applied to the bottom chamber as a chemoattractant [24]. Cell migration was assessed from time 0 through 18 hours by bottom-read fluorescence at 525 nm (Tecan Infinite M200) with each time point Relative Fluorescent Units (RFU) normalized to time=0 RFU for each respective condition. Net migration was calculated as a fold change of the total migration for each condition over total control (vehicle) migration at 18 hours.
Cell cycle analysis: Cells were plated in 12-well plates at 80,000 cells/well in complete media until ~50% confluent. Cells were quiesced in 0.5% FBS for 24 hours followed by treatment in complete growth media (DMEM, 10% FBS, Primocin) containing select pharmacologic agents for 24 hours. Cells were trypsinized, formalin-fixed and stained with PI (Invitrogen) per manufacturer’s recommendations [9,21]. The fraction of cells present in each phase of the cell cycle was assessed by flow cytometry.
Anti-VASP Lv-shRNA infection: Scrambled, NTC controls or Lv-shRNA (SMART vector 2.0) directed against the full-length VASP (Dharmacon Research, Lafayette, CO) were utilized to reduce expression of endogenous VASP. Infection efficiencies and appropriate multiplicity of infection (MOI) were determined per manufacturer’s guidelines (data not shown). Rat ASM cells were plated in 96-well plates at 40% confluence and switched to low serum (0.5%) following adherence. Cells were treated with Lv-shRNA SMART vector 2.0 directed against VASP (MOI 0.3-5), NTC or vehicle for 24 hours in antibiotic-free, low serum media. Media was changed to high glucose-DMEM, 15% FBS and penicillin/streptomycin mix after 24 hours, and on day 3 cells were expanded to 24-well plates and VASP mutants ((-)VASP cells) were selected by puromycin resistance for 24 hours. Stable cell lines were established and used for verification of VASP deficiency as well as for cellular signaling and migration and proliferation analyses.
Statistical analyses: Data were analyzed using Excel 2011/2013 (Microsoft) and Sigma Plot 11.2 (SPSS, Inc.). All data sets were tested for normal distribution and met the homogeneity prerequisites for Analysis of Variance (ANOVA). One-way ANOVA and Tukey’s post-hoc multiple comparison tests were used to detect changes between individual groups. Two-way ANOVA with multiple comparisons and Tukey’s post-hoc tests were used for migration and cell cycle analyses to detect significance between groups. Data are expressed as mean ± Standard Error of the Mean (SEM) with p<0.05 considered statistically significant.
RESULTS
AICAR-stimulated AMPK phosphorylates VASP at select Ser/Thr residues
Previous work from our lab established an AICAR dosing regimen that significantly induces AMPK expression and activity in rat primary and A7r5 ASM cells [9-11]. Following this approach, in the current study we used AICAR to stimulate AMPK signaling and evaluated downstream targets including VASP in rat A7r5 ASM cell growth. Using immunofluorescence with flow cytometry, AICAR (1mM, 60 min) significantly doubled phosphorylation of protein kinase A-dependent VASPSer157 and AMPK-dependent VASPThr278 normalized to total VASP (Figure 1).There were no observable changes in VASPSer239phosphorylation, previously reported to be PKG-dependent [17,20,23], in AICAR-treated cells compared to vehicle controls.
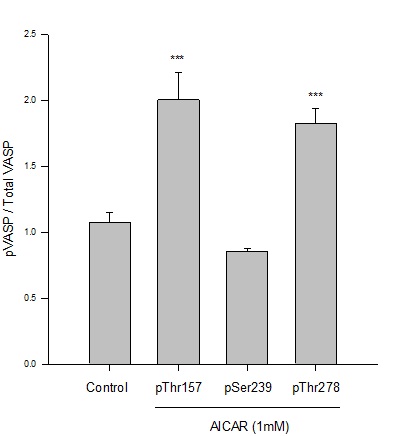
Figure 1: AICAR-stimulated AMPK phosphorylates VASP at specific Ser/Thr sites in ASM cells. Rat A7r5 ASM cells were treated with vehicle (DMSO) or the AMPK stimulator AICAR (1 mM) and site-specific VASP phosphorylation (and total VASP protein) was analyzed by immunofluorescence and flow cytometry after 60 minutes. AMPK significantly increased phosphorylation of VASP at Ser157 and Thr278 (normalized to total VASP) yet did not markedly affect VASP at Ser239 compared to vehicle (DMSO) controls. n=3-5 independent experiments per group; *** = p<0.001 compared to controls.
AICAR-stimulated AMPK promotes cytoskeletal and focal adhesion stability
Results from immunofluorescence and flow cytometry show that AICAR-mediated AMPK (1 mM, 60 min) significantly increased the G:F action ratio (Figure 2), significantly reduced expression of FAKTyr397normalized to total FAK (Figure 3), and significantly increased expression of paxillin (Figure 4) compared to respective vehicle controls.
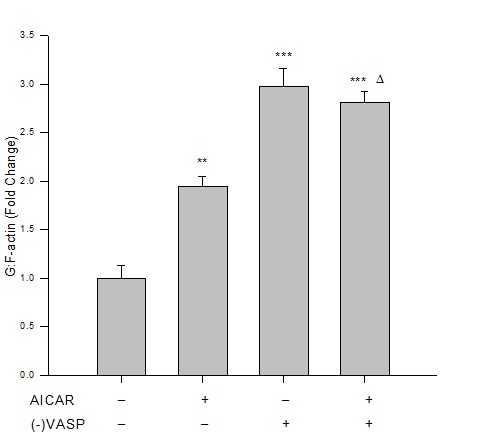
Figure 2: AICAR-stimulated AMPK or VASP deficiency increases G:F action. Control or VASP-deficient ((-)VASP) rat A7r5 ASM cells were treated with vehicle (DMSO) or the AMPK stimulator AICAR (1 mM, 60 min), fixed and stained for G-actin (fluorescently-tagged deoxyribonuclease I) and F-actin (fluorescently-tagged phalloidin), and read by flow cytometry. Immunofluorescence reveals that AICAR promotes F-actin disassembly and/or enhances G-actin stability as shown by significantly increased G:F action ratios compared to vehicle controls. Lv-shRNA-mediated (-)VASP further increased G:F-actin, yet no differences were detected when (-)VASP cells were treated with AICAR. Data are presented as fold change of the total G-actin to F-actin fluorescence detected by flow cytometry. n=3-5 independent experiments per group. ** = p<0.01 compared to controls; ***= p<0.001 compared to controls; ? = p<0.05 compared to AICAR alone.
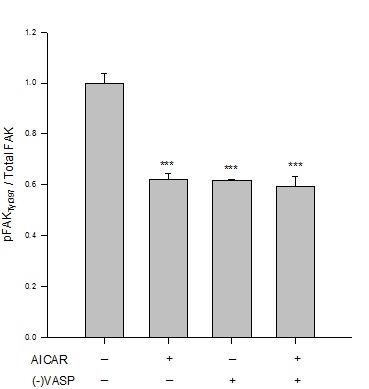
Figure 3: AICAR-stimulated AMPK/VASP inhibits FAK activation. Control or VASP-deficient ((-)VASP) rat A7r5 ASM cells were treated with vehicle (DMSO) or the AMPK stimulator AICAR (1 mM, 60 min) and FAKTyr397 phosphorylation was measured by flow cytometry as an indication of kinase activity [22]. AICAR-treated cells showed significant reduction in FAK activation as measured by FAKTyr397phosphorylation (normalized to total FAK) compared to vehicle controls. Notably, (-)VASP cells also showed significantly reduced FAK activity alone (basally) or in the presence of the AMPK stimulator AICAR. Data are presented as FAKTyr397/total FAK detected by flow cytometry. n=3-5 individual experiments per group. *** = p<0.001 compared to vehicle controls.
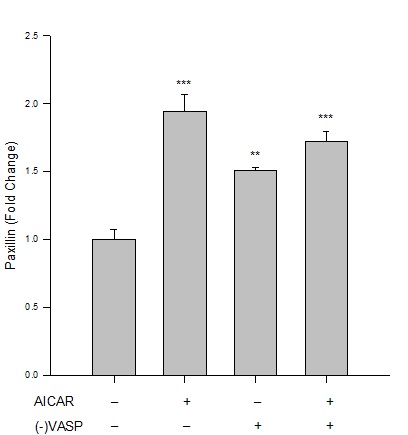
Figure 4: AICAR-stimulated AMPK/VASP promotes focal adhesion stability. Control or VASP-deficient ((-)VASP) rat A7r5 ASM cells were treated with vehicle (DMSO) or the AMPK stimulator AICAR (1 mM, 60 min) and paxillin expression was analyzed by immunolabeling and flow cytometry. Protein analysis reveals that AICAR-stimulated AMPK increases paxillin expression compared to vehicle controls, indicative of focal adhesion stability and anti-migratory signaling. (-)VASP ASM cells alone (basally) or in the presence of AICAR showed significant elevation of paxillin expression compared to controls (although no statistical differences were detected between these two groups). n=3-5 independent experiments per group. ** = p<0.01 compared to controls; *** = p<0.001 compared to controls.
AICAR-stimulated AMPK reduces ASM cell migration and cell cycle progression
Using a modified Boyden transwell chemotactic assay, AICAR-induced AMPK (1 mM) significantly inhibited PDGF-stimulated (10 ng/ml) ASM cell migration compared to vehicle controls after 18 hours (Figure 5). Using flow cytometry for analysis of cell cycle progression, AICAR-stimulated AMPK (1 mM) modestly (p=0.09) elevated cell numbers in G0/G1, significantly increased cell numbers in S, and significantly reduced cell numbers in G2/M compared to vehicle controls after 24 hours (Figure 6).
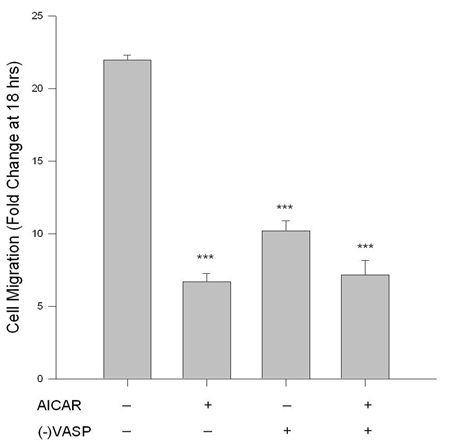
Figure 5: AICAR-stimulated AMPK/VASP inhibits ASM cell migration. Control or VASP-deficient ((-)VASP) rat A7r5 ASM cells were labeled with CellTracker Green and treated with vehicle (DMSO) or the AMPK stimulator AICAR (1 mM), and PDGF-stimulated (10 ng/mL) chemotaxis was evaluated using a modified Boyden transwell chamber and bottom-read fluorescence at 525 nm between 0 and 18 hours. AICAR alone significantly reduced cell migration in ASM cells at 18 hours. (-)VASP ASM cells also showed significant reduction in migration; however, there were no significant differences between (-)VASP cells with or without AICAR. Two-way ANOVA with Tukey’s post-hoc testing was used for multiple comparisons across time and within each treatment group. n=3 independent experiments per group. *** = p<0.001 compared to controls.
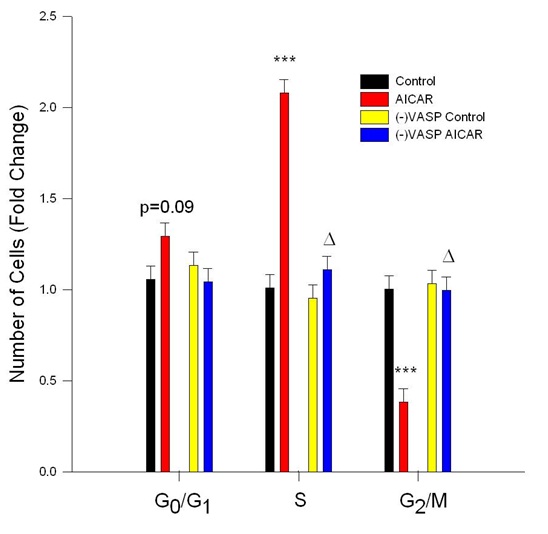
Figure 6: AICAR-stimulated AMPK inhibits cell cycle progression in VASP-dependent manner. Control or VASP-deficient ((-)VASP) rat A7r5 ASM cells were treated with vehicle (DMSO or the AMPK stimulator AICAR (1 mM) for 24 hours following overnight quiescence, and cell cycle progression was analyzed by flow cytometry using Draq5. AICAR alone significantly inhibited cell cycle progression revealed by non-significant increases in G0/G1 phase cells (p=0.09) and significant increases in S-phase cells and significant reduction in G2/M phase cells. (-)VASP reversed the cytostatic effects of AMPK revealed by restoration of the observed increase in S-phase cells and reduction in G2/M phase cells to control levels. n=3 independent experiments per group. *** = p<0.001 compared to controls; ? = p<0.05 compared to AICAR alone.
Generation and validation of Lv-mediated (-)VASP cells
Considering the observed selective, site-specific phosphorylation of VASP and the cytostatic, anti-growth actions following AICAR stimulation of AMPK in ASM cells, we generated VASP-deficient cells using Lv-mediated shRNA directed against full-length VASP ((-)VASP) to test for VASP dependency of these AMPK actions. To validate VASP knockdown, using ECL-based Western blotting on cell homogenates after 24 hours (Figure 7A) and In-Cell Western (ICW) blotting on fixed but intact adherent cells (normalized to DNA within each well) after 48 hours (Figure 7B), results show efficient protein knockdown of VASP in (-)VASP cells compared to scrambled Lv-NTC, vehicle, and empty (no vehicle) controls. Notably, no differences in VASP expression were observed between NTC, vehicle controls, and empty controls using each approach.
Next, we examined VASPSer157 and VASPThr278 phosphorylation in order to confirm site-specific (total and phosphorylated) VASP knockdown in (-)VASP cells in the absence or presence of AICAR stimulation. Using ICW analyses, in the absence of AICAR (-)VASP cells showed significant reduction of phosphorylation of both VASPSer157 and VASPThr278 compared to vehicle controls. Correspondingly, previously observed AICAR-induced increases in phosphorylation of both VASPSer157 and VASPThr278were significantly reduced to below control levels in the VASP-deficient cells compared to vehicle controls (Figures 7C and 7D).
Next, we examined VASPSer157 and VASPThr278 phosphorylation in order to confirm site-specific (total and phosphorylated) VASP knockdown in (-)VASP cells in the absence or presence of AICAR stimulation. Using ICW analyses, in the absence of AICAR (-)VASP cells showed significant reduction of phosphorylation of both VASPSer157 and VASPThr278 compared to vehicle controls. Correspondingly, previously observed AICAR-induced increases in phosphorylation of both VASPSer157 and VASPThr278were significantly reduced to below control levels in the VASP-deficient cells compared to vehicle controls (Figures 7C and 7D).
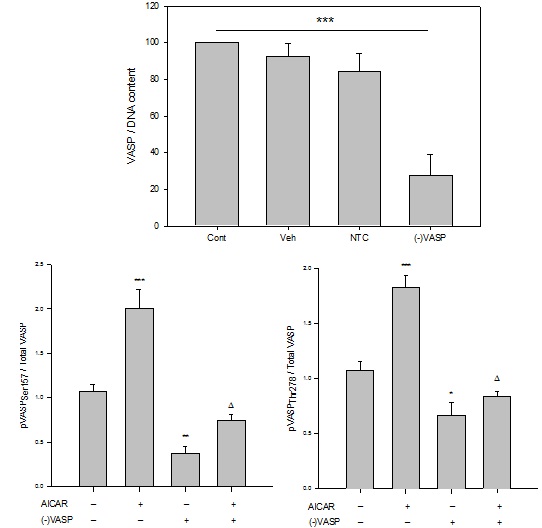
Figure 7: Lentiviral anti-VASP shRNA ((-)VASP) reduces basal and AICAR-stimulated AMPK phosphorylation in ASM cells. Lentiviral shRNA directed against full length VASP ((-)VASP) was generated, and rat A7r5 ASM cells were infected for 60 minutes after which validation of target knockdown was performed in the absence or presence of the AMPK stimulator AICAR.
(A)Western blot for total VASP performed on rat A7r5 ASM cell homogenates after incubation for 24 hours with (-)VASP reveals successful reduction of VASP protein compared to scrambled, non-targeting (NTC) and vehicle (Veh) controls. Lanes under “(-)VASP” labelled “A”, “B” and “C” correspond to samples from three replicates of anti-VASPLv-shRNA-treated cells to show consistency of target ablation, and Veh is the vehicle control (DMEM, 15% FBS, penicillin/streptomycin).
(B) Graphical representation of results obtained from In-Cell Western (ICW) blotting for total VASP (normalized to DNA content within each well) on intact, adherent A7r5 ASM cells after 48 hours incubation with Veh, NTC, or (-)VASP. Significant changes were not observed between Veh, NTC and empty controls (Cont); however, three (-)VASP cell cohorts show significant and consistent knockout of total VASP protein compared to NTC, Veh and Cont controls.
(C) and (D) Site-specificity of (-)VASP following incubation of rat A7r5 ASM cells with anti-VASP Lv-shRNA after 48 hours in the absence or presence of AMPK stimulation by AICAR. Treatment of cells with AICAR (1 mM) stimulated phosphorylation of VASPSer157 (C) and VASPThr278 (normalized to total VASP) (D), respectively, compared to vehicle controls. (-)VASP cells show reduced basal and AMPK-stimulated levels of VASPSer157 and VASPThr278 (normalized to total VASP), respectively. Data are presented as pVASP/total VASP. n=3-5 independent experiments per group.*= p<0.05 compared to controls; **= p<0.01 compared to controls; ***= p<0.001 compared to controls; ? = p<0.05 compared to AICAR alone.
(-)VASP increases G:F-actin ratios, reduces FAKTyr397, and increases paxillin
Under basal (non-AMPK-stimulated) conditions, (-)VASP cells displayed significantly greater G:F actin ratios (Figure 2), significantly reduced FAKTyr397 phosphorylation (Figure 3), and significantly increased expression of paxillin (Figure 4) compared to respective vehicle controls cells. In the presence of AICAR, (-)VASP cells showed significantly greater G:F actin ratios compared to cells treated with AICAR alone (Figure 2). No significant differences were observed in the degree of FAKTyr397 phosphorylation (Figure 3) or in paxillin expression (Figure 4) in AICAR-treated (-)VASP cells compared to cells treated with AICAR alone.
(-)VASP reduces cell migration and reverses AICAR-mediated cytostasis
Under basal conditions cell migration was significantly reduced in (-)VASP cells compared to vehicle control cells after 18 hours (Figure 5). No significant differences were observed in the degree of cell migration in the (-)VASP cells in the presence or absence of AICAR.
Notably, under basal conditions VASP deficiency failed to markedly alter cell cycle progression compared to vehicle controls (Figure 6), yet in the presence of AICAR VASP deficiency completely reversed cytostasis in the S and G2/M phases observed with AICAR alone. No significant differences were observed in cell cycle progression in the (-)VASP cells in the presence or absence of AICAR.
Notably, under basal conditions VASP deficiency failed to markedly alter cell cycle progression compared to vehicle controls (Figure 6), yet in the presence of AICAR VASP deficiency completely reversed cytostasis in the S and G2/M phases observed with AICAR alone. No significant differences were observed in cell cycle progression in the (-)VASP cells in the presence or absence of AICAR.
DISCUSSION
Findings presented in this study support our hypothesis that the metabolic regulator AMPK inhibits ASM cell growth through mechanisms dependent at least in part on the actin cytoskeletal proteinVASP. Using rat A7r5 ASM cells, results show that AICAR-stimulated AMPK phosphorylates select Ser/Thr residues on VASP, stabilizes actin dynamics and focal adhesion/cytoskeletal proteins, and reduces migration and proliferation. Through use of Lv-shRNA-mediated knockdown, results also suggest that AMPK operates to control ASM cell proliferation, at least partly, in VASP-dependent manner. These findings complement and extend some recent studies from our lab [9,10] by providing support for VASP as a novel mechanism underlying anti-growth actions of AMPK and lend credence for consideration of VASP as a suitable biological marker for detection and perhaps prevention of AMPK-related metabolic and cardiovascular disorders.
AMPK has long been studied as a regulator of metabolism and energy balance, and recent findings suggest that AMPK also plays significant roles in the control of cellular growth [2,6-8]. In rat primary ASM, we previously showed that AMPK, stimulated by the AMP mimetic and AMPK stimulator AICAR or the non-metabolic, AMPK-activating small molecule A-769662, communicates with protein kinase A and Ser/Thr phosphatases to reduce migration and proliferation through mechanisms involving the cytoskeletal and focal adhesion factors paxillin and FAK, the matrix-degrading metalloproteinase MMP-9 and its tissue inhibitor TIMP-1, and VASP [9,10]. These anti-migratory and anti-proliferative effects of AMPK are consistent with the findings of others in human and rodent ASM [25,26]. Moreover, complementing its growth inhibitory actions AMPK has been shown to elicit vascular relaxation [6,27] and to maintain vascular barrier function during hypoxia [28], thus making it an attractive candidate for further study in the vasculature.
To advance our understanding and to better define the role of AMPK in the growth regulation of ASM, and considering earlier observations implicating VASP in the mechanisms of AMPK, in the current study we investigated VASP dependency and cytoskeletal and focal adhesion involvement in AMPK-mediated growth inhibition of ASM cells. VASP is implicated in the directionality of extra- to intra-cellular signaling events and in the regulation of focal adhesions and, via nucleotide exchange factor regulation, in the control of transcriptional activation [16,19,29]. Additionally, we recently showed that AMPK has capacity to control differential VASP phosphorylation in the growth inhibition of rat primary ASM [9,10]. In the current study in A7r5 ASM cells we examined induction of site-specific VASP phosphorylation by AICAR-stimulated AMPK. Indeed, AMPK induced significant increases in VASPSer157 and VASPThr278 with little effect on VASPSer239, all compared to vehicle (DMSO) controls (Figure 1). Site-specific phosphorylation of VASPSer157 and VASPSer239 have been used as respective markers of active PKA and PKG signaling [17,20-22], and in fact we recently reported that AMPK operates via crosstalk with PKA and induces VASPSer157phosphorylation to elicit growth control in primary ASM cells [9], supporting the current observations. Phosphorylation at VASPThr278, an AMPK-sensitive site [23], has also been previously reported by our group in primary tissues following AMPK stimulation [10], and along with VASPSer157 can serve to cap actin filaments and prevent polymerization, thus potentially operating as an anti-migratory element. To our knowledge, this is the first report demonstrating stimulation of VASP and its site-specific phosphorylation by AMPK in rat A7r5 ASM cells, a widely used and valuable experimental model with which to investigate cellular growth and matrix/cytoskeletal involvement [22,30].
The actin cytoskeleton acts as an anchoring point for focal adhesion and cell attachment; therefore, with decreased actin polymerization (increased G:F actin) less force will be exerted on focal adhesions and in turn, directed filament elongation required for lamellipodia and filopodia formation (and ensuing cell migration) will be minimized. Since VASP acts as an anti-capping protein associated with polymerizing actin filaments, antagonizing normal VASP function through differential phosphorylation at previously reported inhibitory sites (Ser157 or Thr278) should increase cytoplasmic G-actin and reduce filamentous (F) actin pools. In the current study, immunofluorescent flow cytometry shows that AICAR significantly increases G:F actin ratio compared to cells treated with Veh alone (Figure 2). These findings support our earlier observations [10] that AMPK increases cytoplasmic G-actin (evidenced as elevated G:F actin), increases stress fiber formation, and in turn, inhibits actin cytoskeletal reorganization essential for cellular growth in rat primary ASM cells.
Complementing actin biology, FAK-mediated actin dynamics and turnover of focal adhesions are needed for cells to grow and these require catalytic phosphorylation of FAKTyr397 [31]. In turn, reduction in FAK activation is thought to be inhibitory for cell growth [22]. In the current study using flow cytometry, catalytic phosphorylation of FAKTyr397 was significantly reduced after treatment with AICAR compared to vehicle controls (Figure 3). These results support earlier findings in rat primary cells [10] and suggest a functional link between cytoskeletal filament elongation and focal adhesion turnover and its control by AMPK.
As a component of FAK activation, the focal adhesion adapter and actin regulatory protein paxillin becomes phosphorylated which then targets GTPase activity and mediates focal contact release at that site, a required step for cell migration [31]. In primary ASM cell homogenates we have shown that AMPK stimulates membranous and nuclear paxillin and simultaneously reduces cytosolic paxillin, indicative of an anti-growth, anti-migratory phenotype [10]. In the current study using immunolabelling and flow cytometry we show that AMPK significantly induces paxillin expression in ASM cell homogenates (Figure 4), agreeing with these earlier findings in primary cells and supporting the notion of AMPK contributing to stability of focal adhesion complexes and anti-growth signaling. Collectively, these data on the actin cytoskeleton (G:F actin) and focal contact proteins FAK and paxillin reveal a discrete signaling network elicited by AMPK, which ultimately leads to inhibition of actin filament elongation and focal adhesion turnover.
As a functional, growth-dependent readout of these biochemical relationships, A7r5 ASM cells were exposed to PDGF-mediated chemotaxis using a transwell migration assay over 18 hours in the absence or presence of AICAR. Results show that with AMPK stimulation, migration of cells was significantly reduced after 18 hours compared to vehicle controls (Figure 5), supporting our earlier anti-migratory observations with AMPK stimulation in rat primary ASM [9-11]. As complement to cell migration, in this study we also tested the ability of AMPK to inhibit cell growth and cell cycle progression. Using flow cytometry in quiesced A7r5 ASM cells, AICAR markedly reduced cell cycle progression after 24 hours as illustrated by moderately increased in cells in G0/G1, a doubling of cells in S phase, and a ~60% reduction in cells in G2/M (Figure 6). These findings in A7R5 ASM cells agree with those observed in our earlier studies using AICAR-treated primary ASM cells [9-11], and together with the migration data provide further support for capacity of AMPK to control growth of ASM cells likely through inhibitory phosphorylation events leading to reduced microfilament elongation and focal complex strain.
Based on evidence for site-specific phosphorylation of VASP and its implication in growth control in A7r5 ASM cells by AMPK as presented here and supported by our earlier observations in primary cells [9,10], we theorized that VASP may be an essential component in the mechanism of AMPK. In turn, we generated VASP-deficient ASM cells ((-)VASP) using Lv-shRNA targeted against full-length VASP (Figure 7). Successful viral infection and knockdown of VASP was demonstrated by probing for total VASP using both ECL-based Western blotting on cell homogenates (Figure 7A) and ICW blotting on adherent, intact cells (normalized to DNA within each well) compared to scrambled NTC and Veh controls (Figure 7B). Validation of VASP knockdown was also examined using site-specific VASP phosphorylation under basal or AMPK-stimulated conditions (Figures 7C and 7D). As expected, under basal (non-stimulated) conditions (-)VASP cells showed significantly reduced phosphorylation of Ser157 and Thr278 compared to controls, and following AICAR treatment (-)VASP cells also showed significant reduction in phosphorylation at both sites compared to cells treated with AICAR alone. These findings validate the efficacy of VASP knockdown following anti-VASP Lv-shRNA infection in rat A7r5 ASM cells.
Following generation of (-)VASP cells we then tested for VASP-dependency of the cell cytoskeletal and growth responses previously observed with AMPK stimulation. (-)VASP cells demonstrated elevated levels of G:F actin compared to Veh controls as well as in the presence of AICAR compared to AICAR treatment alone. This is not surprising, considering VASP as a cytoskeletal protein operates in a pro-polymerization manner, and so when VASP gets phosphorylated by AMPK the G-actin pool and thus, G:F actin, should increase (and in turn, relative F-actin should decrease). Next, considering that a proposed mechanism of FAKTyr397 autophosphorylation, an event necessary for kinase activation and focal adhesion turnover, is F-actin strain on focal adhesion complexes [29,31-33], it has been suggested that VASP may play a role in adhesion-directed actin polymerization [29,33]; therefore, the interaction of AMPK with VASP may subsequently reduce FAK auto-activation as a component of its mechanism of action. Interestingly, loss of VASP failed to markedly alter FAKTyr397 (normalized to total FAK) with/without the presence of AICAR. (-)VASP cells also demonstrated significantly elevated paxillin expression in the absence or presence of AICAR compared to controls (Figure 4).
If VASP inhibition reduces actin polymerization and subsequent actin strain on focal adhesions, then inhibiting normal VASP function, either through phosphorylation or genetic knock down, should result in functional inhibition of cellular migration. We recently showed that AMPK mediates inhibition of rat primary ASM cell migration [9-11], and in the current study when (-)VASP ASM cells were exposed to PDGF stimulation, significant reduction in cell migration was also observed yet without significant differences in the absence or presence of AICAR (Figure 5). Lastly, we tested the influence of AMPK with/without VASP ablation on A7r5 ASM cell proliferation. Although (-)VASP cells alone showed similar levels of cell cycle progression compared to control cells, when (-)VASP cells were treated with AICAR a complete reversal of the cytostatic effects of AMPK were observed in all phases of the cell cycle (Figure 6). While the exact mechanism of AMPK-mediated, VASP-dependent inhibition of cell cycle progression remains unclear, it has been suggested that VASP is necessary for Rho-dependent, serum response element (Sre)-mediated transcriptional activity [29,34]. Additionally, it has been suggested that AMPK exhibits inhibitory crosstalk with RhoA in ASM cells [35]. Given this insight and the new evidence presented here that AMPK exerts regulatory impact on VASP, we can speculate that AMPK inhibits ASM cell proliferation via inhibitory regulation of VASP, possibly through VASP-dependent Sre transcriptional activity; however, further investigation into these exact mechanisms are needed. Overall, these data provide evidence that AMPK-mediated ASM cellgrowth is (at least partly) dependent upon cytoskeletal protein VASP.
Bioutility of VASP in AMPK-Moderated Vascular Disease
As mentioned, VASP has long been used as an indirect readout of kinase activity, and only recently has its phosphorylation status been leveraged to independently predict cellular growth phenotype [36]. In this work Storz and colleagues [36] characterized a correlation between breast cancer aggressiveness, protein kinase D2 (PKD2) expression, and VASP phosphorylation at Ser322. They found a negative correlation between PKD2 expression and VASPSer322 with regards to breast cancer aggressiveness which was similarly linked to Relapse-Free Survival (RFS) in patients [36]. These findings from the cancer field provide a rational link between the results presented in this study where we evaluated AMPK and VASPSer157 and VASPThr278 in ASM cells.. Speculatively our data suggest measuring VASP phosphorylation levels and perhaps AMPK activity in CVD patients could similarly predict the aggressiveness of a vascular occlusion. Further we have elucidated a novel and tangible pharmacotherapeutic target in AMPK-mediated VASP phosphorylation which might prove capable of preventing uncontrolled vascular growth and which may then serve as a plausible biologically-relevant marker for AMPK-mediated vascular disorders.
The schematic in Figure 8 depicts a proposed central role for VASP in AMPK-mediated control of ASM cell growth based on these findings in rat A7r5 ASM cells. Through phosphorylation of VASP at Ser157 and Thr278, AMPK exerts inhibitory control which reduces cytoskeletal elongation and relieves focal adhesion strain. These events result in stabilized focal adhesion complexes which, in turn, reduce the ability of cells to proliferate and migrate. Interestingly, following VASP knockdown the influence of AMPK on cell migration was unchanged, indicating that VASP alone has capacity to regulate migration and chemotaxis irrespective of AMPK signaling. Conversely, VASP knockdown completely reversed the inhibitory effects of AMPK on cell proliferation, suggesting a key dependence on AMPK for VASP in its cytostatic effects on ASM cells. While the exact mechanisms of this novel interaction between AMPK and VASP remain incompletely understood, it is intriguing to speculate that AMPK may operate via Rho/Sre in transcriptional control of ASM growth. Alternatively, it is possible that AMPK operates through protein kinase D to elicit VASP phosphorylation at Ser322, a stimulus for intracellular VASP re-localization and a potential new VASP-specific mechanism in the control of vascular growth [36,37]. Collectively, these findings in rat A7r5 ASM cells reveal a possible synergy between AMPK and VASP and suggest the AMPK/VASP nexus as a potential new target for the regulation of pathologic growth associated with ASM-dependent proliferative disorders. Moreover these observations lend credence for the potential bioutility of VASP as a marker of vascular occlusive disorders.
AMPK has long been studied as a regulator of metabolism and energy balance, and recent findings suggest that AMPK also plays significant roles in the control of cellular growth [2,6-8]. In rat primary ASM, we previously showed that AMPK, stimulated by the AMP mimetic and AMPK stimulator AICAR or the non-metabolic, AMPK-activating small molecule A-769662, communicates with protein kinase A and Ser/Thr phosphatases to reduce migration and proliferation through mechanisms involving the cytoskeletal and focal adhesion factors paxillin and FAK, the matrix-degrading metalloproteinase MMP-9 and its tissue inhibitor TIMP-1, and VASP [9,10]. These anti-migratory and anti-proliferative effects of AMPK are consistent with the findings of others in human and rodent ASM [25,26]. Moreover, complementing its growth inhibitory actions AMPK has been shown to elicit vascular relaxation [6,27] and to maintain vascular barrier function during hypoxia [28], thus making it an attractive candidate for further study in the vasculature.
To advance our understanding and to better define the role of AMPK in the growth regulation of ASM, and considering earlier observations implicating VASP in the mechanisms of AMPK, in the current study we investigated VASP dependency and cytoskeletal and focal adhesion involvement in AMPK-mediated growth inhibition of ASM cells. VASP is implicated in the directionality of extra- to intra-cellular signaling events and in the regulation of focal adhesions and, via nucleotide exchange factor regulation, in the control of transcriptional activation [16,19,29]. Additionally, we recently showed that AMPK has capacity to control differential VASP phosphorylation in the growth inhibition of rat primary ASM [9,10]. In the current study in A7r5 ASM cells we examined induction of site-specific VASP phosphorylation by AICAR-stimulated AMPK. Indeed, AMPK induced significant increases in VASPSer157 and VASPThr278 with little effect on VASPSer239, all compared to vehicle (DMSO) controls (Figure 1). Site-specific phosphorylation of VASPSer157 and VASPSer239 have been used as respective markers of active PKA and PKG signaling [17,20-22], and in fact we recently reported that AMPK operates via crosstalk with PKA and induces VASPSer157phosphorylation to elicit growth control in primary ASM cells [9], supporting the current observations. Phosphorylation at VASPThr278, an AMPK-sensitive site [23], has also been previously reported by our group in primary tissues following AMPK stimulation [10], and along with VASPSer157 can serve to cap actin filaments and prevent polymerization, thus potentially operating as an anti-migratory element. To our knowledge, this is the first report demonstrating stimulation of VASP and its site-specific phosphorylation by AMPK in rat A7r5 ASM cells, a widely used and valuable experimental model with which to investigate cellular growth and matrix/cytoskeletal involvement [22,30].
The actin cytoskeleton acts as an anchoring point for focal adhesion and cell attachment; therefore, with decreased actin polymerization (increased G:F actin) less force will be exerted on focal adhesions and in turn, directed filament elongation required for lamellipodia and filopodia formation (and ensuing cell migration) will be minimized. Since VASP acts as an anti-capping protein associated with polymerizing actin filaments, antagonizing normal VASP function through differential phosphorylation at previously reported inhibitory sites (Ser157 or Thr278) should increase cytoplasmic G-actin and reduce filamentous (F) actin pools. In the current study, immunofluorescent flow cytometry shows that AICAR significantly increases G:F actin ratio compared to cells treated with Veh alone (Figure 2). These findings support our earlier observations [10] that AMPK increases cytoplasmic G-actin (evidenced as elevated G:F actin), increases stress fiber formation, and in turn, inhibits actin cytoskeletal reorganization essential for cellular growth in rat primary ASM cells.
Complementing actin biology, FAK-mediated actin dynamics and turnover of focal adhesions are needed for cells to grow and these require catalytic phosphorylation of FAKTyr397 [31]. In turn, reduction in FAK activation is thought to be inhibitory for cell growth [22]. In the current study using flow cytometry, catalytic phosphorylation of FAKTyr397 was significantly reduced after treatment with AICAR compared to vehicle controls (Figure 3). These results support earlier findings in rat primary cells [10] and suggest a functional link between cytoskeletal filament elongation and focal adhesion turnover and its control by AMPK.
As a component of FAK activation, the focal adhesion adapter and actin regulatory protein paxillin becomes phosphorylated which then targets GTPase activity and mediates focal contact release at that site, a required step for cell migration [31]. In primary ASM cell homogenates we have shown that AMPK stimulates membranous and nuclear paxillin and simultaneously reduces cytosolic paxillin, indicative of an anti-growth, anti-migratory phenotype [10]. In the current study using immunolabelling and flow cytometry we show that AMPK significantly induces paxillin expression in ASM cell homogenates (Figure 4), agreeing with these earlier findings in primary cells and supporting the notion of AMPK contributing to stability of focal adhesion complexes and anti-growth signaling. Collectively, these data on the actin cytoskeleton (G:F actin) and focal contact proteins FAK and paxillin reveal a discrete signaling network elicited by AMPK, which ultimately leads to inhibition of actin filament elongation and focal adhesion turnover.
As a functional, growth-dependent readout of these biochemical relationships, A7r5 ASM cells were exposed to PDGF-mediated chemotaxis using a transwell migration assay over 18 hours in the absence or presence of AICAR. Results show that with AMPK stimulation, migration of cells was significantly reduced after 18 hours compared to vehicle controls (Figure 5), supporting our earlier anti-migratory observations with AMPK stimulation in rat primary ASM [9-11]. As complement to cell migration, in this study we also tested the ability of AMPK to inhibit cell growth and cell cycle progression. Using flow cytometry in quiesced A7r5 ASM cells, AICAR markedly reduced cell cycle progression after 24 hours as illustrated by moderately increased in cells in G0/G1, a doubling of cells in S phase, and a ~60% reduction in cells in G2/M (Figure 6). These findings in A7R5 ASM cells agree with those observed in our earlier studies using AICAR-treated primary ASM cells [9-11], and together with the migration data provide further support for capacity of AMPK to control growth of ASM cells likely through inhibitory phosphorylation events leading to reduced microfilament elongation and focal complex strain.
Based on evidence for site-specific phosphorylation of VASP and its implication in growth control in A7r5 ASM cells by AMPK as presented here and supported by our earlier observations in primary cells [9,10], we theorized that VASP may be an essential component in the mechanism of AMPK. In turn, we generated VASP-deficient ASM cells ((-)VASP) using Lv-shRNA targeted against full-length VASP (Figure 7). Successful viral infection and knockdown of VASP was demonstrated by probing for total VASP using both ECL-based Western blotting on cell homogenates (Figure 7A) and ICW blotting on adherent, intact cells (normalized to DNA within each well) compared to scrambled NTC and Veh controls (Figure 7B). Validation of VASP knockdown was also examined using site-specific VASP phosphorylation under basal or AMPK-stimulated conditions (Figures 7C and 7D). As expected, under basal (non-stimulated) conditions (-)VASP cells showed significantly reduced phosphorylation of Ser157 and Thr278 compared to controls, and following AICAR treatment (-)VASP cells also showed significant reduction in phosphorylation at both sites compared to cells treated with AICAR alone. These findings validate the efficacy of VASP knockdown following anti-VASP Lv-shRNA infection in rat A7r5 ASM cells.
Following generation of (-)VASP cells we then tested for VASP-dependency of the cell cytoskeletal and growth responses previously observed with AMPK stimulation. (-)VASP cells demonstrated elevated levels of G:F actin compared to Veh controls as well as in the presence of AICAR compared to AICAR treatment alone. This is not surprising, considering VASP as a cytoskeletal protein operates in a pro-polymerization manner, and so when VASP gets phosphorylated by AMPK the G-actin pool and thus, G:F actin, should increase (and in turn, relative F-actin should decrease). Next, considering that a proposed mechanism of FAKTyr397 autophosphorylation, an event necessary for kinase activation and focal adhesion turnover, is F-actin strain on focal adhesion complexes [29,31-33], it has been suggested that VASP may play a role in adhesion-directed actin polymerization [29,33]; therefore, the interaction of AMPK with VASP may subsequently reduce FAK auto-activation as a component of its mechanism of action. Interestingly, loss of VASP failed to markedly alter FAKTyr397 (normalized to total FAK) with/without the presence of AICAR. (-)VASP cells also demonstrated significantly elevated paxillin expression in the absence or presence of AICAR compared to controls (Figure 4).
If VASP inhibition reduces actin polymerization and subsequent actin strain on focal adhesions, then inhibiting normal VASP function, either through phosphorylation or genetic knock down, should result in functional inhibition of cellular migration. We recently showed that AMPK mediates inhibition of rat primary ASM cell migration [9-11], and in the current study when (-)VASP ASM cells were exposed to PDGF stimulation, significant reduction in cell migration was also observed yet without significant differences in the absence or presence of AICAR (Figure 5). Lastly, we tested the influence of AMPK with/without VASP ablation on A7r5 ASM cell proliferation. Although (-)VASP cells alone showed similar levels of cell cycle progression compared to control cells, when (-)VASP cells were treated with AICAR a complete reversal of the cytostatic effects of AMPK were observed in all phases of the cell cycle (Figure 6). While the exact mechanism of AMPK-mediated, VASP-dependent inhibition of cell cycle progression remains unclear, it has been suggested that VASP is necessary for Rho-dependent, serum response element (Sre)-mediated transcriptional activity [29,34]. Additionally, it has been suggested that AMPK exhibits inhibitory crosstalk with RhoA in ASM cells [35]. Given this insight and the new evidence presented here that AMPK exerts regulatory impact on VASP, we can speculate that AMPK inhibits ASM cell proliferation via inhibitory regulation of VASP, possibly through VASP-dependent Sre transcriptional activity; however, further investigation into these exact mechanisms are needed. Overall, these data provide evidence that AMPK-mediated ASM cellgrowth is (at least partly) dependent upon cytoskeletal protein VASP.
Bioutility of VASP in AMPK-Moderated Vascular Disease
As mentioned, VASP has long been used as an indirect readout of kinase activity, and only recently has its phosphorylation status been leveraged to independently predict cellular growth phenotype [36]. In this work Storz and colleagues [36] characterized a correlation between breast cancer aggressiveness, protein kinase D2 (PKD2) expression, and VASP phosphorylation at Ser322. They found a negative correlation between PKD2 expression and VASPSer322 with regards to breast cancer aggressiveness which was similarly linked to Relapse-Free Survival (RFS) in patients [36]. These findings from the cancer field provide a rational link between the results presented in this study where we evaluated AMPK and VASPSer157 and VASPThr278 in ASM cells.. Speculatively our data suggest measuring VASP phosphorylation levels and perhaps AMPK activity in CVD patients could similarly predict the aggressiveness of a vascular occlusion. Further we have elucidated a novel and tangible pharmacotherapeutic target in AMPK-mediated VASP phosphorylation which might prove capable of preventing uncontrolled vascular growth and which may then serve as a plausible biologically-relevant marker for AMPK-mediated vascular disorders.
The schematic in Figure 8 depicts a proposed central role for VASP in AMPK-mediated control of ASM cell growth based on these findings in rat A7r5 ASM cells. Through phosphorylation of VASP at Ser157 and Thr278, AMPK exerts inhibitory control which reduces cytoskeletal elongation and relieves focal adhesion strain. These events result in stabilized focal adhesion complexes which, in turn, reduce the ability of cells to proliferate and migrate. Interestingly, following VASP knockdown the influence of AMPK on cell migration was unchanged, indicating that VASP alone has capacity to regulate migration and chemotaxis irrespective of AMPK signaling. Conversely, VASP knockdown completely reversed the inhibitory effects of AMPK on cell proliferation, suggesting a key dependence on AMPK for VASP in its cytostatic effects on ASM cells. While the exact mechanisms of this novel interaction between AMPK and VASP remain incompletely understood, it is intriguing to speculate that AMPK may operate via Rho/Sre in transcriptional control of ASM growth. Alternatively, it is possible that AMPK operates through protein kinase D to elicit VASP phosphorylation at Ser322, a stimulus for intracellular VASP re-localization and a potential new VASP-specific mechanism in the control of vascular growth [36,37]. Collectively, these findings in rat A7r5 ASM cells reveal a possible synergy between AMPK and VASP and suggest the AMPK/VASP nexus as a potential new target for the regulation of pathologic growth associated with ASM-dependent proliferative disorders. Moreover these observations lend credence for the potential bioutility of VASP as a marker of vascular occlusive disorders.
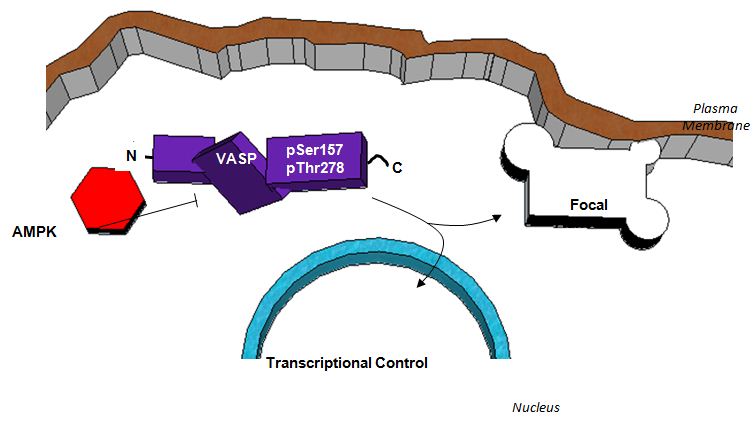
Figure 8: Schematic depicting a proposed functional relationship between AMPK and VASP in the inhibition of ASM cell growth. AMPK exhibits inhibitory control of VASP through phosphorylation which reduces directed microfilament elongation and relieves focal adhesion strain. A reduction in focal adhesion strain results in reduced auto phosphorylation of focal adhesion kinase (FAK) and subsequent activation of accessory focal adhesion proteins. Increased stability of focal adhesions ultimately reduces membrane detachment from the extracellular matrix which is required for cell movement. Additionally, VASP inhibition reduces microfilament elongation necessary for lamellipodia and filopodia formation also required for cell movement and proliferation. Finally, VASP inhibition appears to reverse the observed AMPK-mediated cytostatic effect on ASM cells. While the exact mechanism remains uncertain, it is clear that AMPK operates to inhibit cell cycle progression and cell proliferation of rat A7r5 ASM cells via regulation of VASP.
ACKNOWLEDGEMENTS
This project was supported by Award Number R01HL081720 from the National Heart, Lung, and Blood Institute, National Institutes of Health (DAT), a Pre-doctoral Fellowship from the American Heart Association (JDS), an ECU Brody School of Medicine Seed/Bridge Grant (DAT), and a Brody Brothers Endowment Fund Award (DAT). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute, the National Institutes of Health, or the American Heart Association.
REFERENCES
- Writing Group Members, Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, et al. (2016) Executive Summary: Heart Disease and Stroke Statistics -- 2016 Update: A Report from the American Heart Association. Circulation 133: 447-454.
- Hardie DG (2011) AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev 25: 1895-1908.
- Hardie DG (2007) AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol 8: 774-785.
- Rubin LJ, Magliola L, Feng X, Jones AW, Hale CC (2005) Metabolic activation of AMP kinase in vascular smooth muscle. J Appl Physiol 98: 296-306.
- Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D, et al. (2007) Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J 403: 139-148.
- Ewart MA, Kennedy S (2011) AMPK and vasculoprotection. Pharmacol Ther 131: 242-253.
- Luo Z, Zang M, Guo W (2010) AMPK as a metabolic tumor suppressor: control of metabolism and cell growth. Future Oncol 6: 457-470.
- Mihaylova, MM, Shaw RJ (2011) The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 13: 1016-1023.
- Stone JD, Narine A, Tulis DA (2012) Inhibition of vascular smooth muscle growth via signaling crosstalk between AMP-activated protein kinase and cAMP-dependent protein kinase. Front Physio 3: 409.
- Stone JD, Narine A, Shaver PR, Fox JC, Vuncannon JR, et al. (2013) AMP-activated protein kinase inhibits vascular smooth muscle cell proliferation and migration and vascular remodeling following injury. Am J Physiol Heart Circ Physiol 304: 369-381.
- Stone JD, Holt AW, Vuncannon JR, Brault JJ, Tulis DA (2015) AMP-activated protein kinase inhibits transforming growth factor-β-mediated vascular smooth muscle cell growth: implications for a Smad-3-dependent mechanism. Am J Physiol Heart Circ Physiol 309: 1251-1259.
- Lund N, Henrion D, Tiede P, Ziche M, Schunkert H, et al. (2010) Vimentin expression influences flow dependent VASP phosphorylation and regulates cell migration and proliferation. Biochem Biophys Res Commun 395: 401-406.
- Holt AW, Martin DN, Shaver PR, Adderley SP, Stone JD, et al. (2016) Soluble guanylyl cyclase-activated cyclic GMP-dependent protein kinase inhibits arterial smooth muscle cell migration independent of VASP-Serine 239 phosphorylation. Cell Signal 28: 1364-1379.
- Barzik M, Kotova TI, Higgs HN, Hazelwood L, Hanein D, et al. (2005) Ena/VASP proteins enhance actin polymerization in the presence of barbed end capping proteins. J Biol Chem 280: 28653-28662.
- Bear JE, Svitkina TM, Krause, M, Schafer DA, Loureiro JJ, et al. (2002) Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell 109: 509-521.
- Breitsprecher D, Kiesewetter AK, Linkner J, Urbanke C, Resch GP, et al. (2008) Clustering of VASP actively drives processive, WH2 domain-mediated actin filament elongation. EMBO J 27: 2943-2954.
- Benz PM, Blume C, Seifert S, Wilhelm S, Waschke J, et al. (2009) Differential VASP phosphorylation controls remodeling of the actin cytoskeleton. J Cell Sci 122: 3954-3965.
- Chen L, Daum G, Chitaley K, Coats SA, Bowen-Pope DF, et al. (2004) Vasodilator-stimulated phosphoprotein regulates proliferation and growth inhibition by nitric oxide in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 24: 1403-1408.
- Worth DC, Hodivala-Dilke K, Robinson SD, King SJ, Morton PE, et al. (2010) Alpha v beta3 integrin spatially regulates VASP and RIAM to control adhesion dynamics and migration. J Cell Biol 189: 369-383.
- Adderley SP, Joshi CN, Martin DN, Tulis DA (2012) Phosphodiesterases regulate BAY 41-2272-induced VASP phosphorylation in vascular smooth muscle cells. Front Pharmacol 3: 10.
- Joshi CN, Martin DN, Fox JC, Mendelev NN, Brown TA, et al. (2011) The soluble guanylate cyclase stimulator BAY 41-2272 inhibits vascular smooth muscle growth through the cAMP-dependent protein kinase and cGMP-dependent protein kinase pathways. J Pharmacol Exp Ther 339: 394-402.
- Mendelev NN, Williams VS, Tulis DA (2009) Antigrowth properties of BAY 41-2272 in vascular smooth muscle cells. J Cardiovasc Pharm 53: 121-131.
- Blume C, Benz PM, Walter U, Ha J, Kemp BE, et al. (2007) AMP-activated protein kinase impairs endothelial actin cytoskeleton assembly by phosphorylating vasodilator-stimulated phosphoprotein. J Biol Chem 282: 4601-4612.
- Yang X, Thomas DP, Zhang X, Culver BW, Alexander BM, et al. (2006) Curcumin inhibits platelet-derived growth factor-stimulated vascular smooth muscle cell function and injury-induced neointima formation. Arterioscler Thromb Vasc Biol 26: 85-90.
- Nagata D, Takeda R, Sata M, Satonaka H, Suzuki E, et al. (2004) AMP-activated protein kinase inhibits angiotensin II-stimulated vascular smooth muscle cell proliferation. Circulation 110: 444-451.
- Igata M, Motoshima H, Tsuruzoe K, Kojima K, Matsumura T, et al. (2005) Adenosine monophosphate-activated protein kinase suppresses vascular smooth muscle cell proliferation through the inhibition of cell cycle progression. Circ Res 97: 837-844.
- Rossoni LV, Wareing M, Wenceslau CF, Al-Abri M, Cobb C, et al. (2011) Acute simvastatin increases endothelial nitric oxide synthase phosphorylation via AMP-activated protein kinase and reduces contractility of isolated rat mesenteric resistance arteries. Clin Sci (Lond) 121: 449-458.
- Schmit MA, Mirakaj V, Stangassinger M, König K, Köhler D, et al. (2012) Vasodilator phosphostimulated Protein (VASP) protects endothelial barrier function during hypoxia. Inflammation 35: 566-573.
- Zhuang S, Nguyen GT, Chen Y, Gudi T, Eigenthaler M, et al. (2004) Vasodilator-stimulated phosphoprotein activation of serum-response element-dependent transcription occurs downstream of RhoA and is inhibited by cGMP-dependent protein kinase phosphorylation. J Biol Chem 279: 10397-10407.
- Murphy DA, Courtneidge SA (2011) The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol 12: 413-426.
- Tomar A, Schlaepfer DD (2009) Focal adhesion kinase: switching between GAPs and GEFs in the regulation of cell motility. Curr Opin Cell Biol 21: 676-683.
- Serrels B, Serrels A, Brunton VG, Holt M, McLean GW (2007) Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nat Cell Biol 9: 1046-1056.
- Friedland JC, Lee MH, Boettiger D (2009) Mechanically activated integrin switch controls alpha5beta1 function. Science 323: 642-644.
- Grosse R, Copeland JW, Newsome TP, Way M, Treisman R (2003) A role for VASP in RhoA-Diaphanous signalling to actin dynamics and SRF activity. EMBO J 22: 3050-3061.
- Gayard M, Guilluy C, Rousselle A, Viollet B, Henrion D, et al. (2011) AMPK alpha 1-induced RhoA phosphorylation mediates vasoprotective effect of estradiol. Arterioscler Thromb Vasc Biol 31: 2634-2642.
- Döppler H, Bastea L, Borges S, Geiger X, Storz P (2015) The phosphorylation status of VASP at serine 322 can be predictive for aggressiveness of invasive ductal carcinoma. Oncotarget 6: 29740-29752.
- Döppler HR, Bastea LI, Lewis-Tuffin LJ, Anastasiadis PZ, Storz P (2013) Protein kinase D1-mediated phosphorylations regulate Vasodilator-Stimulated Phosphoprotein (VASP) localization and cell migration. J Biol Chem 288: 24382-24393.
Citation: Stone JD, Holt AW, Shaver PR, Vuncannon JR, Tulis DA (2016) AMP-Activated Protein Kinase Inhibits Arterial Smooth Muscle Cell Proliferation in Vasodilator-Stimulated Phosphoprotein-Dependent Manner. J Non Invasive Vasc Invest 1: 002.
Copyright: © 2016 David A Tulis, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
© 2024, Copyrights Herald Scholarly Open Access. All Rights Reserved!