Journal of Nephrology & Renal Therapy Category: Clinical
Type: Case Report
Antineutrophil Cytoplasmic Antibody Pauci-Immune Crescentic Glomerulonephritis and Interstitial Lung Disease in Scleroderma. Case Report and Literature Review
*Corresponding Author(s):
Leslie Dubin KerrDepartment Of Medicine And Geriatrics, Icahn School Of Medicine At Mount Sinai, Box 1244, One Gustave L. Levy Place, New York, NY 10029, United States
Tel:+1 2122416065,
Fax:+1 2129875584
Email:leslie.kerr@mssm.edu
Received Date: Mar 25, 2016
Accepted Date: Apr 29, 2016
Published Date: May 16, 2016
Abstract
We present 3 cases of acute kidney injury in patients with scleroderma complicated by interstitial lung disease. Scleroderma renal crisis was initially suspected, but upon further evaluation all patients had documented antineutrophil cytoplasmic antibody associated glomerulonephritis. The first patient was treated with cyclophosphamide followed by rituximab with improvement in creatinine and is maintained on mycophenolate mofetil. The second patient, incidentally found to have a creatinine of 11 mg/dl, was treated with pulse steroids and oral cyclophosphamide. She is currently on hemodialysis. The third patient presented with an acute pulmonary renal syndrome. He initially responded but died 2 months later from septicemia. We also present a review of the published literature from 1994 to 2015. Patients were mostly female with limited scleroderma, prolonged duration of scleroderma symptoms prior to acute kidney injury, interstitial lung disease, positive anti-topoisomerase 1 and anti-myeloperoxidase antibodies. Antineutrophil cytoplasmic antibody associated glomerulonephritis should be included in the differential of scleroderma with interstitial lung disease and acute kidney injury.
Keywords
Antineutrophil Cytoplasmic; Lung Disease
INTRODUCTION
Scleroderma is a systemic autoimmune disease with heterogeneous clinical manifestations. Patients demonstrate varying degrees of skin thickening, Raynaud’s phenomenon, nail fold capillary abnormalities, GERD, dysphagia, small oral aperture, telangiectasia, and calcinosis. Internal organ complications include scleroderma renal crisis, interstitial lung disease, pulmonary hypertension and gastrointestinal dysmotility. Scleroderma renal crisis is characterized by hypertension, Acute Kidney Injury (AKI) and microangiopathic hemolytic anemia [1]. Acute kidney injury in the setting of scleroderma is generally assumed to reflect scleroderma renal crisis which occurs in 5-10% of patients with scleroderma [2,3]. Less commonly diagnosed in these patients is Pauci-immune Crescentic Glomerulonephritis (PICGN) as a cause of acute renal failure. We report 3 cases of myeloperoxidase Antineutrophil Cytoplasmic Antibody (ANCA) associated biopsy proven PICGN in patients with diffuse scleroderma. A review of the previously published literature from 1994 to 2015 is presented here to emphasize to physicians this diagnostic possibility.
CASE SERIES
Case 1
A 60-year-old female with scleroderma (gastroesophageal reflux (GERD), Raynaud’s, fingertip pitting scars, diffuse hand edema, periungal capillary changes, mildly decreased oral aperture, ANA 1:640 and anti-topoisomerase I of 8.0, normal range < 1.0 IU) and usual interstitial pneumonia diagnosed on lung biopsy 4 years earlier was readmitted to another hospital after treatment of pneumonia 1 month prior. Broncho-alveolar lavage was reported as negative for Pneumocystis, fungi or malignant cells. She reported elevated creatinine and blood pressure but exact values are unavailable. Urinalysis results were not available to us. Scleroderma renal crisis was considered the likely diagnosis, however, after serology was positive for myeloperoxidase-ANCA a kidney biopsy was performed. Final diagnosis was changed to ANCA PICGN. She previously failed treatment with high dose steroids and Mycophenolate Mofetil (MMF) for pulmonary symptoms and most recently had received 4 doses of monthly intravenous cyclophosphamide prior to succumbing to pneumonia. Treatment with four doses of weekly rituximab 375 mg/m2 was initiated. After four years of follow up her creatinine is stable at 1.4 mg/dl; baseline creatinine was 1.2 mg/dl. Two years ago right heart catheterization demonstrated pulmonary hypertension and her chest CT this year has worsened slightly from the initial CT 5 years ago which reported Interstitial Lung Disease (ILD) with extensive honey-combing and associated traction bronchiectatic changes most prominent at the lung bases. She is currently maintained on MMF at 2 g/day.
Case 2
A 41-year-old female was admitted for AKI found incidentally during pre-operative assessment for right heart catheterization to investigate pulmonary hypertension. She had an 8-year history of idiopathic pulmonary fibrosis, diagnosed on lung biopsy. She also reported a 10-year history of Raynaud’s, a 6-year history of dysphagia, GERD, a noticeably smaller oral aperture, rash, alopecia and joint pains. Examination was notable for a small oral aperture, tight skin on her face, moderate bilateral pitting edema, bibasilar crepitations and lack of sclerodactyly. While her admission blood pressure of 100/70 was only marginally higher than her baseline of 90/60; her creatinine was elevated at 11.3 mg/dl, with an active urine sediment (protein, erythrocytes, leukocytes). Serology was notable for ANA 1:320 and repeatedly negative anti-centromere and anti-topoisomerase I. Scleroderma renal crisis was considered and hemodialysis was initiated along with Angiotensin Converting Enzyme (ACE) inhibitors and calcium channel blockers. Right heart catheterization revealed moderate pulmonary hypertension and increased pulmonary capillary wedge pressures (previous transthoracic echocardiogram revealed an EF of 18%). A diagnosis of myocarditis secondary to diffuse scleroderma was considered. Workup revealed positive myeloperoxidase-ANCA. Hematoxylin and eosin stains of the renal biopsy demonstrated acute, organized fibrocellular crescents. Immunofluorescence was negative and electron microscopy failed to reveal immune type deposits. There were no histological changes to the arteries or arterioles. A diagnosis of PICGN was reported. The patient received 3 days of pulse steroids along with oral daily cyclophosphamide. Her renal function did not recover and she continued on hemodialysis after discharge. While her ejection fraction improved and pulmonary hypertension normalized her exercise tolerance has recently decreased and she is being investigated for worsening interstitial lung disease.
Case 3
The patient, a 55-year-old male with a 30-year history of diffuse scleroderma, presented to the inpatient service after a 2-year absence from medical care with acute shortness of breath and hemoptysis. Scleroderma diagnosis was based on Raynaud’s, sclerodactyly, digital ulcers, dysphagia and interstitial lung disease. Serology results were notable for ANA 1:160 and negative anti-topoisomerase 1. Previous treatments included D-penicillamine but no recent immunosuppression. Admission blood pressure was elevated to 140/71 and urinalysis revealed protein, leukocytes and numerous erythrocytes but no casts. Admission creatinine was 1.7 mg/dl, eGFR 49 ml/min. A renal biopsy was performed on day 4 of admission for worsening creatinine. Hematoxylin and eosin stains revealed intraglomerular fibrinoid necrosis, cellular crescents, endocapillary and extracapillary proliferation, acute tubular necrosis and erythrocyte casts. Immunofluorescence was positive for granular staining of the mesangium and capillary loops with IgG (3+), IgA (2+), C3 (2+), C1q (2+), kappa (2+) and lambda (2+). There was also granular staining of the tubular basement membranes and peritubular capillaries with IgG. Electron microscopy demonstrated mainly subepithelial and intra-membranous deposits rather than subendothelial deposits. The degree of necrosis and crescent formation significantly exceeded that of endocapillary proliferation or subendothelial deposit formation that is typically seen in lupus nephritis. Also, myeloperoxidase-ANCA returned positive while Ro, La, Smith and anticardiolipin antibodies were negative. Complement levels were normal and lupus anticoagulant and anti-dsDNA antibodies were reported as equivocal. With this information the patient’s pathology findings were classified as PICGN with class II and class V lupus nephritis without changes consistent with scleroderma renal crisis. Notably, there was no history of photosensitivity, pleuritic chest pain, oral ulcers, patchy hair loss or small joint arthralgia. He was started on methylprednisolone 1g daily along with IV cyclophosphamide. The patient’s oxygen requirements continued to improve after 7 sessions of plasma exchange, oral daily cyclophosphamide and tapering doses of steroids. He was eventually discharged home on 4 L/min nasal cannula oxygen and a creatinine of 1.9 mg/dl; baseline 1.6 mg/dl. Two months after discharge he succumbed to E. coli septicemia.
LITERATURE REVIEW
We present a PubMed search for English language publications between 1994 and 2015 using combinations of the search terms “scleroderma”, “systemic sclerosis”, “antineutrophil cytoplasmic antibodies” “pauci-immune glomerulonephritis”, “crescentic glomerulonephritis”, “glomerulonephritis”, “acute kidney injury”, “myeloperoxidase” and “microscopic polyangiitis”. Additionally, the references of the studies identified were then examined for further reports. Clinical features (gender, age at scleroderma and glomerulonephritis diagnosis, presence of hemoptysis, previous D penicillamine use, blood pressure, skin involvement) and laboratory data (proteinuria, creatinine, serology, renal biopsy pathology) were captured from case reports. The presence of ILD was also noted. Prescribed treatments and renal and mortality outcomes were recorded.
A total of 29 reports with 47 cases were published (Table 1) [4-32]. We included 8 cases with a history of D-penicillamine use. Six cases were excluded: 1 case complicated by renal cell cancer [33], a case of arthralgia and GERD but without Raynaud’s or skin changes to suggest scleroderma [24], a report of vasculitis of the foot without renal involvement [16], and 3 cases where the renal biopsy findings were not typical of PICGN (extensive deposition of IgG, IgA, IgM and C3 on immunofluorescence [34], renal biopsy describing glomerular sclerosis rather than Glomerulonephritis (GN) [35], and a renal biopsy with the overall impression of crescentic GN with granular immune deposits [36]).
A total of 29 reports with 47 cases were published (Table 1) [4-32]. We included 8 cases with a history of D-penicillamine use. Six cases were excluded: 1 case complicated by renal cell cancer [33], a case of arthralgia and GERD but without Raynaud’s or skin changes to suggest scleroderma [24], a report of vasculitis of the foot without renal involvement [16], and 3 cases where the renal biopsy findings were not typical of PICGN (extensive deposition of IgG, IgA, IgM and C3 on immunofluorescence [34], renal biopsy describing glomerular sclerosis rather than Glomerulonephritis (GN) [35], and a renal biopsy with the overall impression of crescentic GN with granular immune deposits [36]).
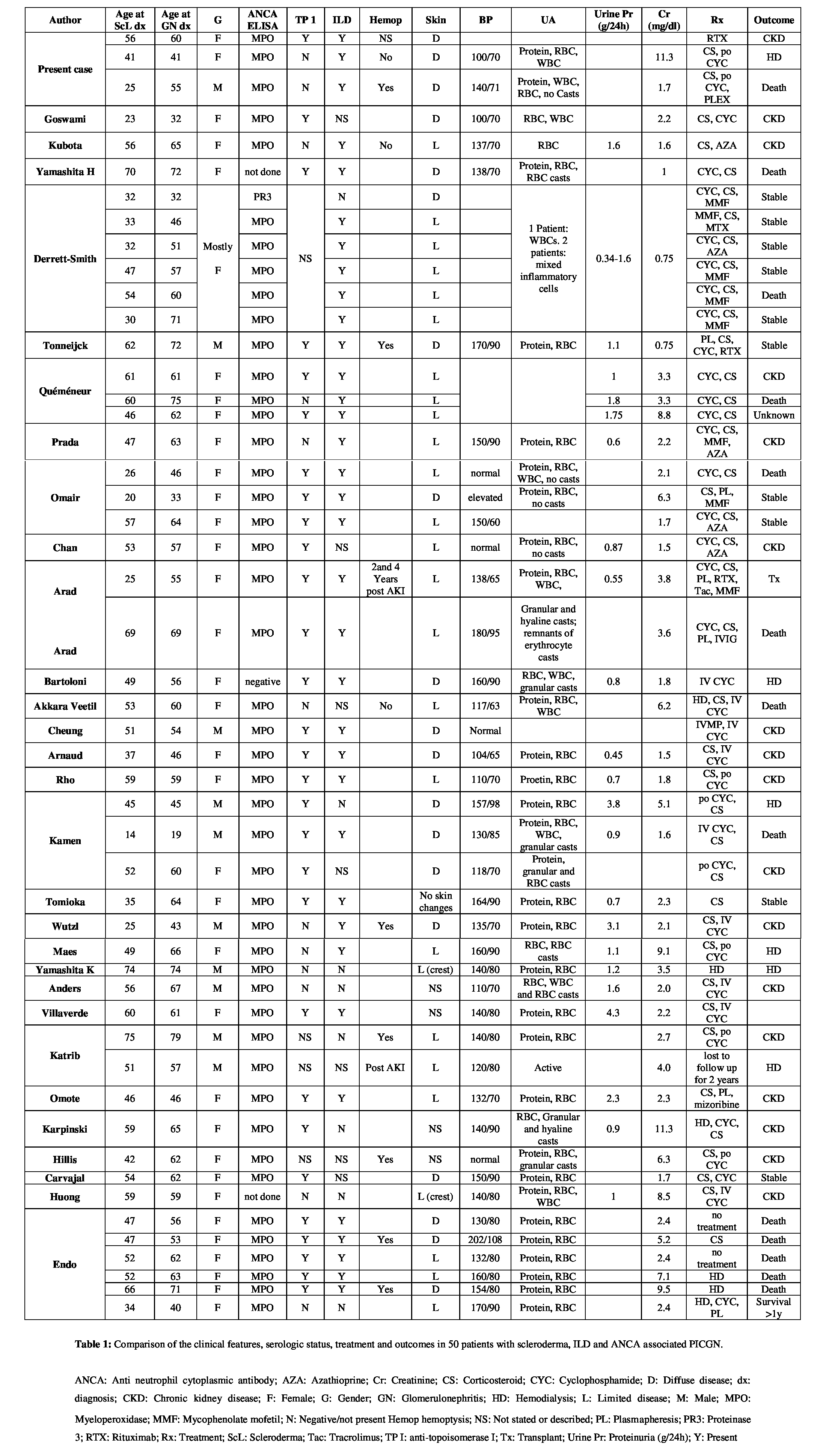
An analysis of the literature including our report of 3 cases reveals 80% of patients were female with a mean age of 57 years at onset of glomerulonephritis after a 10-year history of scleroderma. Of the 50 patients, 26 had limited disease, 19 diffuse disease and in 5 cases the description of skin changes did not allow classification. Thirty-five patients were reported to have interstitial lung disease. Seven patients had hemoptysis at time of acute kidney injury and 2 patients after the episode of glomerulonephritis. Blood pressure values were available for 35 patients with a mean systolic of 140 mmHg with 8 patients having a systolic greater than or equal to 160 mmHg. Urinalysis on 45 patients revealed almost all had hematuria and proteinuria. Erythrocyte casts were reported in 6 cases and granular casts in a similar number. Quantification of proteinuria was reported in 28 patients: values ranged from 0.3 to 4.3 g/day, mean 1.4 g/day. Creatinine averaged 3.8 mg/dl from 47 patients at diagnosis of AKI. All patients tested positive for anti-MPO except for 1 positive result for PR3, 1 negative and 2 not tested (92% positive). Of the 41 patients tested for anti-topoisomerase I, 29 were positive resulting in a sensitivity of 71%, similar to the findings by Arad [11] and Rho [16].
With regards to treatment, all patients were prescribed steroids with cyclophosphamide except 9 patients not treated with steroids and 12 not treated with cyclophosphamide. There were 12 deaths, 5 from a report by Endo in 1994. Seven patients required long term hemodialysis or renal transplant, 18 patients were left with some renal dysfunction and in 11 patients creatinine returned to normal. Outcomes were not reported in 2 patients.
A recent review in 2012 by Chan and Mok of 19 patients with PICGN in scleroderma revealed similar findings [10]. Their review excluded drug induced cases. Patients were mostly female in the 6th decade of life. While our review demonstrated a slightly higher percentage of patients with limited disease there were equal numbers of limited and diffuse disease in the review by Chan. Eighty-two percent of patients had a blood pressure less than 160 mmHg systolic, similar to our finding, and 10% had nephrotic range proteinuria. Ninety-five percent of patients were MPO-ANCA positive. Roughly 2/3 of patients developed end stage renal disease and the mortality rate was 36%.
DISCUSSION
ANCA antibodies have been reported in association with connective tissue diseases, most commonly systemic lupus erythematosus with prevalence rates of 16% to 31% [37-39]. Double positivity has also been described in patients who have both ANCA and anti-GBM antibodies [40]. Despite prior reports of ANCA associated PIGN in scleroderma in the literature, we believe clinicians are still unaware of this possibility especially in patients with coexisting ILD. Thus ANCAs are rarely drawn and patients are treated as scleroderma renal crisis with Angiotensin Converting Enzyme (ACE) inhibitors only. The treatment of PICGN requires high doses of steroids combined with immunosuppression rather than ACE inhibitors. This series again emphasizes the importance of considering this diagnostic possibility in any scleroderma patient with ILD in whom AKI is developing.
Our cases share some features typical of ANCA associated PICGN in scleroderma patients reported in the literature. The prolonged duration of scleroderma symptoms prior to kidney injury is consistent with the report by Arad et al., [11]. Theses authors compared scleroderma renal crisis with ANCA associated PICGN in scleroderma patients; median duration of symptoms in scleroderma renal crisis was 7 months versus a median of 7 years in scleroderma with ANCA renal disease. Generally, there is a female predominance, with onset of kidney injury in the 5th to 6th decade of life. The majority of patients have interstitial lung disease and positive anti-topoisomerase 1. ANCA glomerulonephritis is not exclusive to either limited or diffuse disease. All our cases occurred in patients with diffuse scleroderma but in our literature review limited skin involvement was described in half of the patients.
The prevalence of ANCA antibodies in scleroderma was previously described by Rufatti et al., in 2002 [41]. In a cohort of 115 patients, 5 tested positive for pANCA with 3 demonstrating antibodies to MPO. While the relationship between ANCA antibodies and glomerulonephritis has been established with animal models the evidence of their involvement in pulmonary fibrosis is less clear [42,43]. In 2011 Guilpain published in vitro evidence supporting anti MPO induced increases in reactive oxygen species [44]. Previous work by Servettaz demonstrated increased fibroblast proliferation with sera from patients with scleroderma and interstitial lung disease that contained high amounts of advanced oxidation protein products [45].
Our cases share some features typical of ANCA associated PICGN in scleroderma patients reported in the literature. The prolonged duration of scleroderma symptoms prior to kidney injury is consistent with the report by Arad et al., [11]. Theses authors compared scleroderma renal crisis with ANCA associated PICGN in scleroderma patients; median duration of symptoms in scleroderma renal crisis was 7 months versus a median of 7 years in scleroderma with ANCA renal disease. Generally, there is a female predominance, with onset of kidney injury in the 5th to 6th decade of life. The majority of patients have interstitial lung disease and positive anti-topoisomerase 1. ANCA glomerulonephritis is not exclusive to either limited or diffuse disease. All our cases occurred in patients with diffuse scleroderma but in our literature review limited skin involvement was described in half of the patients.
The prevalence of ANCA antibodies in scleroderma was previously described by Rufatti et al., in 2002 [41]. In a cohort of 115 patients, 5 tested positive for pANCA with 3 demonstrating antibodies to MPO. While the relationship between ANCA antibodies and glomerulonephritis has been established with animal models the evidence of their involvement in pulmonary fibrosis is less clear [42,43]. In 2011 Guilpain published in vitro evidence supporting anti MPO induced increases in reactive oxygen species [44]. Previous work by Servettaz demonstrated increased fibroblast proliferation with sera from patients with scleroderma and interstitial lung disease that contained high amounts of advanced oxidation protein products [45].
CONCLUSION
We present three cases of scleroderma and interstitial lung disease who had an acute decline in kidney function. While scleroderma renal crisis was initially considered, testing for anti-myeloperoxidase antibody followed by visualization of PICGN on biopsy confirmed an ANCA vasculitis which had important implications for management. Suspicion for PICGN in scleroderma should be higher in patients with long symptom duration, lack of severe hypertension, interstitial lung disease and positive anti-topoisomerase 1. While we are unable to comment on treatment efficacy given the limited number of cases, recognition of PICGN enables effective treatment with cyclophosphamide or rituximab.
REFERENCES
- Guillevin L, Mouthon L (2015) Scleroderma Renal Crisis. Rheum Dis Clin North Am 41: 475-488.
- Penn H, Howie AJ, Kingdon EJ, Bunn CC, Stratton RJ, et al. (2007) Scleroderma renal crisis: patient characteristics and long-term outcomes. QJM 100: 485-494.
- Denton CP, Lapadula G, Mouthon L, Müller-Ladner U (2009) Renal complications and scleroderma renal crisis. Rheumatology (Oxford) 48: 32-35.
- Yamashita H, Takahashi Y, Kaneko H, Kano T, Mimori A (2014) A patient with diffuse cutaneous systemic sclerosis complicated by antineutrophil-cytoplasmic antibody-associated vasculitis exhibiting honeycomb lung without volume loss. Intern Med 53: 801-804.
- Derrett-Smith EC, Nihtyanova SI, Harvey J, Salama AD, Denton CP (2013) Revisiting ANCA-associated vasculitis in systemic sclerosis: clinical, serological and immunogenetic factors. Rheumatology (Oxford) 52:1824-1831.
- Tonneijck L, Tanna A, Pusey CD (2013) Antineutrophil cytoplasm antibody-positive pulmonary-renal syndrome in a patient with diffuse cutaneous systemic sclerosis. BMJ Case Rep.
- Quéméneur T, Mouthon L, Cacoub P, Meyer O, Michon-Pasturel U, et al. (2013) Systemic vasculitis during the course of systemic sclerosis: report of 12 cases and review of the literature. Medicine (Baltimore) 92: 1-9.
- Zurita Prada PA, Martín Rodríguez S, Urrego Laurín CL, Heras Benito M, Molina Ordas A, et al. (2013) ANCA vasculitis in a patient with systemic sclerosis. Reumatol Clin 9: 72-73.
- Omair MA, Mohamed N, Johnson SR, Ahmad Z, Lee P (2013) ANCA-associated vasculitis in systemic sclerosis report of 3 cases. Rheumatol Int 33: 139-143.
- Chan PT, Mok CC (2012) Pauci-immune crescentic glomerulonephritis in limited cutaneous systemic sclerosis. Clin Rheumatol 31: 1273-1277.
- Arad U, Balbir-Gurman A, Doenyas-Barak K, Amit-Vazina M, Caspi D, et al. (2011) Anti-neutrophil antibody associated vasculitis in systemic sclerosis. Semin Arthritis Rheum 41: 223-229.
- Bartoloni Bocci E, Giordano A, Luccioli F, Alunno A, Gerli R (2009) Crescentic glomerulonephritis and circulating antineutrophil cytoplasmic antibodies in scleroderma: a case report and review of the literature. Clin Exp Rheumatol 27: 385-386.
- Akkara Veetil BM, Schimmer BM (2009) A case of limited systemic sclerosis with p-ANCA, complicated by multiple cerebral hemorrhages. Rheumatol Int 29: 325-329.
- Cheung G, Chew G, Wyndham R, Peters M, Riminton S (2007) Myeloperoxidase-antineutrophil cytoplasmic antibody seroconversion and fulminant vasculitis in Scl-70-positive scleroderma. Intern Med J 37: 205-207.
- Arnaud L, Huart A, Plaisier E, Francois H, Mougenot B, et al. (2007) ANCA-related crescentic glomerulonephritis in systemic sclerosis: revisiting the “normotensive scleroderma renal crisis”. Clin Nephrol 68: 165-170.
- Rho YH, Choi SJ, Lee YH, Ji JD, Song GG (2006) Scleroderma associated with ANCA-associated vasculitis. Rheumatol Int 26: 369-375.
- Kamen DL, Wigley FM, Brown AN (2006) Antineutrophil cytoplasmic antibody-positive crescentic glomerulonephritis in scleroderma--a different kind of renal crisis. J Rheumatol 33: 1886-1888.
- Tomioka M, Hinoshita F, Miyauchi N, Akiyama Y, Saima S, et al. (2004) ANCA-related crescentic glomerulonephritis in a patient with scleroderma without marked dermatological change and malignant hypertension. Intern Med 43: 496-502.
- Wutzl AL, Foley RN, O’Driscoll BR, Reeve RS, Chisholm R, et al. (2001) Microscopic polyangiitis presenting as pulmonary-renal syndrome in a patient with long-standing diffuse cutaneous systemic sclerosis and antibodies to myeloperoxidase. Arthritis Rheum 45: 533-536.
- Maes B, Van Mieghem A, Messiaen T, Kuypers D, Van Damme B, et al. (2000) Limited cutaneous systemic sclerosis associated with MPO-ANCA positive renal small vessel vasculitis of the microscopic polyangiitis type. Am J Kidney Dis 36: 16.1-16.5.
- Yamashita K, Yorioka N, Kyuden Y, Naito T, Tanji C, et al. (2000) A case of CREST syndrome and myeloperoxidase-specific anti-neutrophil cytoplasmic autoantibody-associated glomerulonephritis. Clin Nephrol 53: 296-300.
- Anders HJ, Wiebecke B, Haedecke C, Sanden S, Combe C, et al. (1999) MPO-ANCA-Positive crescentic glomerulonephritis: a distinct entity of scleroderma renal disease? Am J Kidney Dis 33: 3.
- Villaverde V, Balsa A, Cabezas JA, Fernández-Prada M, Torre A, et al. (1999) Normotensive renal failure in a patient with systemic sclerosis and p-antineutrophil cytoplasmic autoantibodies which developed into Paget’s disease of bone after immunosuppressive therapy. Rheumatology (Oxford) 38: 190-191.
- Katrib A, Sturgess A, Bertouch JV (1999) Systemic sclerosis and antineutrophil cytoplasmic autoantibody-associated renal failure. Rheumatol Int 19: 61-63.
- Omote A, Muramatsu M, Sugimoto Y, Hosono S, Murakami R, et al. (1997) Myeloperoxidase-specific anti-neutrophil cytoplasmic autoantibodies -- related scleroderma renal crisis treated with double-filtration plasmapheresis. Intern Med 36: 508-513.
- Karpinski J, Jothy S, Radoux V, Levy M, Baran D (1997) D-penicillamine-induced crescentic glomerulonephritis and antimyeloperoxidase antibodies in a patient with scleroderma. Case report and review of the literature. Am J Nephrol 17: 528-532.
- Hillis GS, Khan IH, Simpson JG, Rees AJ (1997) Scleroderma, D-penicillamine treatment, and progressive renal failure associated with positive antimyeloperoxidase antineutrophil cytoplasmic antibodies. Am J Kidney Dis 30: 279-281.
- Carvajal I, Bernis C, Sanz P, Garcia A, Garcia-Vadillo A, et al. (1997) Antineutrophil Cytoplasmic Autoantibodies (ANCA) and systemic sclerosis. Nephrol Dial Transplant 12: 576-577.
- Huong DL, Papo T, Gatfosse M, Frances C, Godeau P, et al. (1995) Antineutrophil cytoplasmic autoantibodies in systemic sclerosis with renal failure. J Rheumatol 22: 791-792.
- Endo H, Hosono T, Kondo H (1994) Antineutrophil cytoplasmic autoantibodies in 6 patients with renal failure and systemic sclerosis. J Rheumatol 21: 864-870.
- Kubota K, Ueno T, Mise K, Hazue R, Suwabe T, et al. (2015) ANCA-Associated Vasculitis in a Patient with Systematic Sclerosis and Sjögren’s Syndrome: A Case Report. Case Rep Nephrol Dial 5: 113-117.
- Goswami RP, Mondal S, Basu K, Das S, Ghosh P, et al. (2015) Acute kidney injury in a patient with systemic sclerosis: Looking beyond scleroderma renal crisis. Indian J Nephrol 25: 387-388.
- Abrich V, Duvuru S, Swanson HJ (2013) Limited scleroderma with pauci-immune glomerulonephritis in the presence of renal cell carcinoma. Clin Med Res 11: 117-119.
- Herrera-Esparza R, Aguilar JL, Saucedo A, González I, López-Robles E, et al. (2005) Scleroderma with type III glomerulonephritis and MPO-ANCA antibodies in the serum. J Eur Acad Dermatol Venereol 19: 617-620.
- Akimoto S, Ishikawa O, Tamura T, Miyachi Y (1996) Antineutrophil cytoplasmic autoantibodies in patients with systemic sclerosis. Br J Dermatol 134: 407-410.
- Ramaswami A, Kandaswamy T, Rajendran T, Jeyakrishnan KP, Aung H, et al. (2008) Scleroderma with crescentic glomerulonephritis: a case report. J Med Case Rep 2: 151.
- Wang Y, Huang X, Cai J, Xie L, Wang W, et al. (2016) Clinicopathologic Characteristics and Outcomes of Lupus Nephritis With Antineutrophil Cytoplasmic Antibody: A Retrospective Study. Medicine (Baltimore) 95: 2580.
- Merkel PA, Polisson RP, Chang Y, Skates SJ, Niles JL (1997) Prevalence of antineutrophil cytoplasmic antibodies in a large inception cohort of patients with connective tissue disease. Ann Intern Med 126: 866-873.
- Galeazzi M, Morozzi G, Sebastiani GD, Bellisai F, Marcolongo R, et al. (1998) Anti-neutrophil cytoplasmic antibodies in 566 European patients with systemic lupus erythematosus: prevalence, clinical associations and correlation with other autoantibodies. European Concerted Action on the Immunogenetics of SLE. Clin Exp Rheumatol 16: 541-546.
- Levy JB, Hammad T, Coulthart A, Dougan T, Pusey CD (2004) Clinical features and outcome of patients with both ANCA and anti-GBM antibodies. Kidney Int 66: 1535-1540.
- Ruffatti A, Sinico RA, Radice A, Ossi E, Cozzi F, et al. (2002) Autoantibodies to proteinase 3 and myeloperoxidase in systemic sclerosis. J Rheumatol 29: 918-923.
- Jennette JC, Falk RJ (2014) Pathogenesis of antineutrophil cytoplasmic autoantibody-mediated disease. Nat Rev Rheumatol 10: 463-473.
- Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, et al. (2002) Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest 110: 955-963.
- Guilpain P, Chéreau C, Goulvestre C, Servettaz A, Montani D, et al. (2011) The oxidation induced by antimyeloperoxidase antibodies triggers fibrosis in microscopic polyangiitis. Eur Respir J 37: 1503-1513.
- Servettaz A, Guilpain P, Goulvestre C, Chéreau C, Hercend C, et al. (2007) Radical oxygen species production induced by advanced oxidation protein products predicts clinical evolution and response to treatment in systemic sclerosis. Ann Rheum Dis 66: 1202-1209.
Citation: Mankee A, Kerr LD (2016) Antineutrophil Cytoplasmic Antibody Pauci-Immune Crescentic Glomerulonephritis and Interstitial Lung Disease in Scleroderma. Case Report and Literature Review. J Nephrol Renal Ther 2: 005.
Copyright: © 2016 Leslie Dubin Kerr, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Journal Highlights
© 2026, Copyrights Herald Scholarly Open Access. All Rights Reserved!