Auditory Findings in Kabuki Syndrome: Description of a Case Report
*Corresponding Author(s):
Janaina Patricio De LimaAudiologist-PhD-Researcher, University Clinic Of Navarre, Spain
Email:jpatriciode@unav.es
Abstract
Kabuki Syndrome (KS) is a rare disease characterised by a wide spectrum of clinical features including craniofacial, ophthalmological, auditory, cardiovascular, cognitive and neurological abnormalities, among others. We report the clinical case of a 16-year-old girl who was diagnosed with this syndrome in early childhood and has unilateral severe mixed hearing loss. In order to treat said hearing impairment, the patient received an active middle ear implant which provided her with satisfactory hearing gain and performance, as well as helping her academic, psychological and social development.
Keywords
Active middle ear implant; Hearing; Hearing loss; Hearing aids; Implants; Kabuki syndrome.
Introduction
Kabuki Syndrome (KS) was first described in 1981 by Niikawa et al. [1] and Kuroki et al. [2]. It is a syndrome that falls within the so-called rare diseases, with an incidence of approximately 1 in 32,000 births [3]. Genetically, KS is characterised by a pathogenic variant in heterozygosity of the KMT2D gene, also known as MLL2, or a heterozygous or homozygous variation of KDM6A [4].
In clinical terms, a patient suffering from this syndrome may present with a wide spectrum of features. Craniofacial alterations include elongated palpebral fissures with eversion of the lateral third of the lower eyelid, arched and broad eyebrows, short columella with depressed nasal tip, large ears (prominent or cupped from growth), cleft lip and/or palate, ogival palate and microcephaly [5,6]. Ophthalmological, dental, cardiovascular, gastrointestinal, endocrinological, immunological, auditory, vestibular, neurological, musculoskeletal and cognitive abnormalities may also be present [7-9].
With regards hearing, the most common condition in KS is conductive hearing loss due to otitis media, however inner ear malformations can also occur [10]. Thus, the types of hearing loss are varied and can be conductive, mixed or sensorineural, with varying degrees of intensity. Hearing rehabilitation will depend on the diagnosis and may require the following treatments: hearing aid, cochlear implant, bone conduction implant, active middle ear implant and/or reconstructive ear surgery.
The present article aims to describe a clinical case of KS.
Clinical Case
The patient is a girl born in 2006 (currently 16 years old). She is the second child of a healthy, non-consanguineous couple. At the time of pregnancy, the mother was 32 years old and the father was 37 years old.
Controlled pregnancy with normal progression. Cystic hygroma at 12 weeks with positive nuchal fold (4.6 mm). Preterm delivery at 36 weeks, with adequate weight for gestational age.
From birth, the child presented the following characteristics:
Neonatal hypotonia.
Cardiac alterations with atrial septal defect and aortic coarctation, which were corrected in the perinatal period.
Arnold-Chiari malformation, type 1, with associated hydro syringomyelia, surgically operated with resection of the left cerebellar tonsil at 10 years of age.
Subclinical hypothyroidism since birth, treated from the age of 4 years with Levothyroxine.
Presence of hypoglycaemia up to 3 years of age, with feeding requirements.
Feeding difficulties, with the presence of gastroesophageal reflux in early childhood.
Typical facial features of KS: hypertelorism with everted lower eyelids and large palpebral fissure, short columella with depressed nasal tip and low-set ears.
History of repeated acute otitis media and repeated urinary tract infections with vesical-ureteral reflux. Currently asymptomatic.
Immunodeficiency: IgA and IgG deficiency with reversal of CD4/CD8 ratio.
Mild hyperopia.
Mild intellectual disability.
Delayed psychomotor development.
Hypodontia.
Bifid uvula.
Padded fingertips.
Microcephaly.
MML2 gene disruption (c.14361dup/p.Val4788Serfs*29). De novo mutation.
Hearing loss detected at birth during hearing screening performed by means of optoacoustic emissions assessment. Severe unilateral mixed hearing loss was diagnosed in the left ear (Figure 1), the conductive component of which was caused by a malformation of the ossicular chain, with fixation of the stapes footplate, and a prominent and dehiscent facial nerve found in the second temporal portion. This anomaly was diagnosed during exploratory tympanotomy surgery, having gone unnoticed in the preoperative CT ear study performed with 0.6 mm sections. The right ear showed thresholds within normality. The tympanometry showed type A curves and an absence of the stapedial reflex in both ears.
For the treatment of hearing loss, the fitting of a hearing aid was indicated at age 6 and subsequently an active middle ear implant, model Vibrant SoundBridge (VSB)/ MED-EL, at age 8. Surgery was carried out to perform a vibroplasty with coupling of the floating mass transducer (FMT) to the round window using a silicone coupler.
The external processor was a SAMBA.
The patient wears her prosthesis regularly and presents adequate functional gain in tone audiometry performed in free field (Figure 2). It is interesting to note the significant functional gain obtained for high-pitched tones, with an excess over bone conduction thresholds at frequencies above 1000Hz. The recognition of monosyllabic words was 84% at 65dB, presented in free field in an open context without visual support.
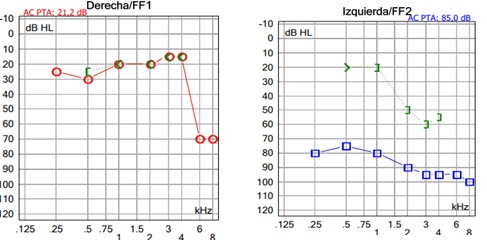 Figure 1: Preoperative liminal tone audiometry.
Figure 1: Preoperative liminal tone audiometry.
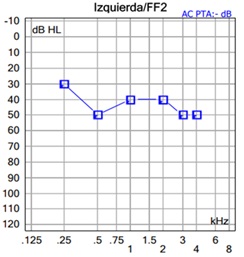 Figure 2: Functional free-field gain with contralateral masking using the Vibrant Sound Bridge implant in the left ear.
Figure 2: Functional free-field gain with contralateral masking using the Vibrant Sound Bridge implant in the left ear.
Discussion
KS is a rare syndrome with wide variations in phenotype expression. The case presented shows the typical clinical features of KS, as reported in the literature. The patient has facial features corresponding to the syndrome and abnormalities in growth and craniofacial development, as well as muscular, cardiac, cognitive, ophthalmological, dermatological, psychomotor and dental abnormalities. In addition, she presented feeding problems and hypothyroidism during childhood. The diagnosis was confirmed by a genetic study, with the confirmation of MML2 gene alteration (de novo mutation).
With regards hearing, the patient has suffered severe unilateral mixed hearing loss since birth due to an ossicular malformation accompanied by a sensory lesion of the cochlea. The indication of treatment for unilateral loss is relatively recent but there is already sufficient evidence of the benefits of restoring a bilateral stimulation of the auditory system [11,12]. Especially in children, unilateral loss can result in poorer language development and academic performance [13]. Furthermore, unilateral loss affects binaural summation and reduces the squelch effect, as well as generating a head "shadow effect" and preventing adequate sound localization [14,15]. Undoubtedly, any intervention must take place early, within the critical time window, in order to avoid an auditory cortical reorganisation that would favour aural dominance [16].
The VSB active middle ear implant chosen to treat this particular case of hearing loss is a semi-implantable prosthesis, which has an external part, where the acoustic signal is captured and converted into an electrical signal, and an internal part, which receives the electrical information and passes it to the FMT. The latter generates a vibratory movement in the structure to which it is attached, such as one of the ossicles, especially the incus and stapes, or the oval and round windows. [17] In this patient, coupling the FMT to the ossicular chain was ruled out due to a fixed stapes foot base. The FMT coupling was performed at the round window level by means of a silicone coupler that makes the energy transfer more efficient. This also provides functional gain in high frequencies with an overlap, necessary in this case as there was a drop in bone conduction thresholds above 1000 Hz, in a context of mixed hearing loss.
The indication for VSB may vary from country to country. In the United States, the indication for active middle ear implantation is restricted to adults, whilst in Europe the indication for children is allowed, provided that the anatomical criteria for surgery are met [18]. Previous studies in adult patients and children with this type of implant show significant hearing gain, improved speech understanding in noisy environments, a high degree of satisfaction in the use of the processor and long-term stability in the results [19-24].
In the KS case presented, the clinical diagnosis preceded the genetic diagnosis, which was confirmatory. Without doubt, we consider that an early genetic diagnosis [25] will contribute positively to speeding up the diagnosis and treatment of children with hearing loss, especially within the context of a syndrome. This syndromic nature requires a multidisciplinary approach. It should be noted that after an 8-year follow-up, the use of the VSB implant was successful, with satisfactory hearing gain and performance which helped the patient in her academic, psychological, and social development.
References
- Niikawa N, Matsuura N, Fukushima Y, Ohsaw T, Kajii T (1981) "Kabuki make-up syndrome: A syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency," J Pediatr. 99: 565-569.
- Kuroki Y, Suzuki Y, Chio H, Hata A, Matsui I (1981) "A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip and skeletal anomalies associated with postnatal dwarfism and mental retardation," J Pediatr. 99: 570-573.
- Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin M (2010) "Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome," Nat Genet. 9: 790-793.
- Adam MP, Banka S, Bjornsson HT, Bodamer O, Chudley AE (2019) "Kabuki syndrome: international consensus diagnostic criteria," J Med Genet. PP: 89-95.
- Adam MP, Hudgins L, Hannibal M (2011) "Kabuki syndrome," GeneReviews, Seattle, WA: Internet.
- Pascual-Castroviejo I, Pacual-Pascual S, Velázquez-Fragua R, Palencia R (2005) "[Kabuki makeup syndrome. A report of 18 Spanish cases]" Rev Naurol. 8: 473-178.
- Digilio M, Gnazzo M, Lepri F, Dentici M, Pisaneschi E (2017) "Congenital heart defects in molecularly proven Kabuki syndrome patients," Am J Med Genet A. PP: 2912-2922.
- Chen Y, Sun M, Hsia S, Lai C, Wu W (2014) "Rare ocular features in a case of Kabuki syndrome (Niikawa-Kuroki syndrome)," BMC Ophthalmol. PP: 143.
- Caciolo C, Alfieri P, Piccini G, Digilio M, Lepri F (2018) "Neurobehavioral features in individuals with Kabuki syndrome," Mol Genet Genomic Med, PP: 322-331.
- Vesseur A, Cillessen E, Mylanus E (2016) "Cochlear Implantation in a Patient with Kabuki Syndrome," J Int Adv Otol, 1: 129-131.
- Nuñez F, Casaubón C, Canet J, Allende A, Zubicaray J (2018) "Diagnosis and early treatment of unilateral or asymmetric hearing loss in childhood: 2017 CODEPEH recommendations," Revista Española de Discapacidad, I: 259-2580.
- Rohlfs A, Friedhoff J, Bohnert A, Breitfuss A, Hess M (2017) "Unilateral hearing loss in children: a retrospective study," Eur J Pedriat. PP: 475-486.
- Fisher C, Lieu J (2014) "Unilateral hearing loss is associated with a negative effect on language," Int J Pediatr Otorhinolaryngol. PP: 1611-1617.
- Kamal S, Robinson A, Diaz R (2012) "Cochlear implantation in single-sided deafness for enhancement of sound localization and speech perception," Curr Opin Otolaryngol Head Neck Surg, PP: 393-397.
- Rana B, Bucholz J, Morgan C, Sharma M, Weller T (2017) "Bilateral Versus Unilateral Cochlear Implantation in Adult Listeners: Speech-On-Speech Masking and Multitalker Localization," Trends in Hearing. 21: 1-15.
- Sharma A, Glick H (2017) "Cross-modal Plasticity in Developmental and Age-Related Hearing Loss: Clinical Implications," Hear Res. 343: 191-201.
- Cochlear WJ (2018) Implants: Audiologic Management and Considerations for Implantable Hearing Devices, Plural Publishing Inc, 2018.
- Cremers C, O'Connor A, Helms J, Roberson J, Clarós P, et al. (2010) "International consensus on Vibrant Soundbridge1 implantation in children and adolescents" Int. J. Pediatr. Otorhinolaryngol. 74: 1267-1269.
- Mandalà M, Colletti L, Colletti V (2011) "Treatment of the Atretic Ear With Round Window Vibrant Soundbridge Implantation in Infants and Children: Electrocochleography and Audiologic Outcomes," Otology & Neurotology, 8: 1250-1255.
- Roman S, Denoyelle F, Farinetti A, Garabedian E, Triglia J (2012) "Middle ear implant in conductive and mixed congenital hearing loss in children," Int. J. Pediatr. Otorhinolaryngol. PP: 1775-1778.
- Lüers J, Hüttenbrink K (2014) "Vibrant Soundbridge Rehabilitation of Conductive and Mixed Hearing Loss," Otoryngol Clin North Am. 47: 915-926.
- Hempel J, Sprinzl G, Riechelmann H, Streitberger C, Giarbini N (2019) "A Transcutaneous Active Middle Ear Implant (AMEI) in Children and Adolescents: Long-term, Multicenter Results" Otology & Neurotology. 8: 1059-1067.
- Takashi M, Iwasaki S, Furutate S, Oka S, Oyamada S (2021) "Active middle ear implant (vibrant soundbridge) in children with unilateral congenital aural atresia," Acta Otolaryngol. 141: 34-38.
- Buhl C, Schindler V, Pfiffner F, Veraguth D, Hube A (2022) "Subjective Sound Quality Detection (HISQUI) over Time after Vibrant Soundbridge Implantation," J Clin Med. 11: 1811.
- Yang T, Guo L, Wang L, Yu X (2019) "Diagnosis, Intervention, and Prevention of Genetic Hearing Loss," in Hearing Loss: Mechanisms, Prevention and Cure. Advances in Experimental Medicine and Biology, 27 ed., Singapore, Springer.
Citation: Lima JP, Manrique M (2024) Auditory Findings in Kabuki Syndrome: Description of a Case Report. J Clin Stud Med Case Rep 11:219
Copyright: © 2024 Janaina Patricio de Lima, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.