Calciphylaxis and Hypotension: Report of a Case and Review of the Literature
*Corresponding Author(s):
Shauna HigginsYale Waterbury Internal Medicine Residency Program, Waterbury, United States
Tel:203573-6162,
Email:shaunahiggins@yale.edu
Abstract
Calciphylaxis, also known as Calcific Uremic Arteriolopathy (CUA), is an often fatal cutaneous disorder of increasing incidence. Its pathogenesis remains unclear and optimal treatment regimens have yet to be clearly outlined. We present a case of a 43-year old male who presented to an outside hospital with severe hypotension of unknown etiology, after which he developed livedo reticularis that progressed to necrosis of the legs, genitalia, and fingers. At UCLA, the patient’s hospital course was complicated by several episodes of hemodynamic instability in the form of Non-sustained Ventricular Tachycardia (NSVT) and episodes of severe hypotension. Following these episodes, the patient’s cutaneous disease was noted to worsen after inter-episode periods of disease stability. In light of the severe mortality associated with a diagnosis of calciphylaxis, any contribution to treatment optimization should be explored. Based on our observations, blood pressure control in the normotensive range may be essential in maintaining disease stability.
Keywords
INTRODUCTION
Calciphylaxis is a relatively rare disorder characterized by diffuse deposition of insoluble calcium salts in the vasculature. Although cutaneous manifestations dominate the clinical presentation, vascular calcifications occur in many vascular beds including those in skeletal muscle, brain, lungs, intestines, eyes, and mesentery [1]. Vascular calcification in the skin leads to ischemia and subsequent cutaneous and subcutaneous necrosis. Clinically it begins as livedo reticularis, a netlike cutaneous discoloration with lighter areas caused by decreased cutaneous blood flow and darker areas caused by impaired dermal plexus venous drainage leading to accumulation of deoxygenated blood. From the initial livedo reticularis stage, calciphylaxis progresses from painful, subcutaneous, purpuric plaques to necrotic ulcers and eschars as continued calcification of blood vessels narrows and increasingly diminishes cutaneous blood and therefore oxygen supply. These large ulcers and eschars represent a severely impaired skin barrier that predispose the calciphylaxis patient to overwhelming sepsis, the predominant cause of mortality [2].
Significant mortality, which nears 80% at one year even with treatment, is due in part to the lack of an optimal or standardized treatment regimen. A multidisciplinary approach is important and multiple interventions have been described, however the overall quality of the evidence is poor [1]. There are no published data from randomized controlled trials that address any of the proposed interventions, thus treatment regimens are based predominantly in expert opinion, clinical experience, and observational published data [1].
Current management of calciphylaxis includes aggressive wound care, hyperbaric oxygen, sodium thiosulfate, and thrombolytic agents. Aggressive wound care includes serial wound debridement, negative pressure wound therapy, and split thickness skin grafts when indicated [1]. Hyperbaric therapy counteracts tissue hypoxia by providing high concentrations of oxygen. It also reduces the risk of infection by providing oxygen necessary for the formation of bactericidal free oxygen radicals [3]. Sodium thiosulfate is the most common therapy and is noted to work as an anticalcification agent by binding calcium and thereby increasing solubility of calcium deposits to enhance their hemodialysis clearance [4]. This is the mechanistic rationale behind administering sodium thiosulfate after dialysis sessions and allowing it to remain in the blood to dissolve calcium deposits until being subsequently removed at the next dialysis session [4]. Recent reports, however, question the calcium-chelating properties of sodium thiosulfate [1]. Instead, they report sodium thiosulfate acts predominantly as an antioxidant and inducer of endothelial nitric oxide synthesis [2,4]. Improvement in pain within one to two weeks of initiating sodium thiosulfate therapy is an important predictor of long-term response [1].
Traditional calciphylaxis therapy also classically addresses the calcium-phosphate-PTH axis by minimizing calcium intake, utilizing calcimimetics, vitamin D analogs, strict phosphate control, and occasional surgical parathyroidectomy when indicated [5]. Serum calcium and phosphorous levels should be maintained in the normal range and serum PTH level should be maintained between 150 and 300 ng/mL with our patient’s being 176 [1]. More novel approaches target alteration of osteoprotegrin and NF-κB pathways and treatment of macrophage and cytokine-mediated inflammation [4]. Although iron has not been definitively proven to be an etiologic agent in development of calciphylaxis, iron products should be stopped nonetheless due to recent reports of its possible role in pathogenesis [6,7]. Targeted molecular therapies may also play a role in treatment or prevention. Two possible molecular targets include Fetuin-A and Matrix Gla, both of which act as vascular calcification inhibitors [3]. Maintenance of hemoglobin between 9.5 and 10.5 g/dL with transfusion rather than erythropoiesis-stimulating agents should also be attempted. Sterile maggot therapy with larvae of the greenbottle fly, Licilia sericata, has also been described as second-line therapy [1]. We present a case of hypotension-associated calciphylaxis, suggesting hemodynamic instability is a pathogenic factor in the development of calciphylaxis and thus demonstrating an important role for blood pressure control in treatment.
REPORT OF A CASE
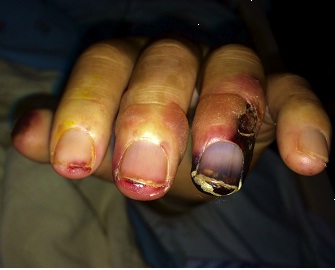
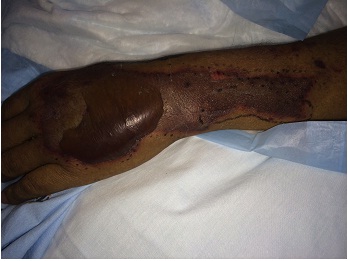
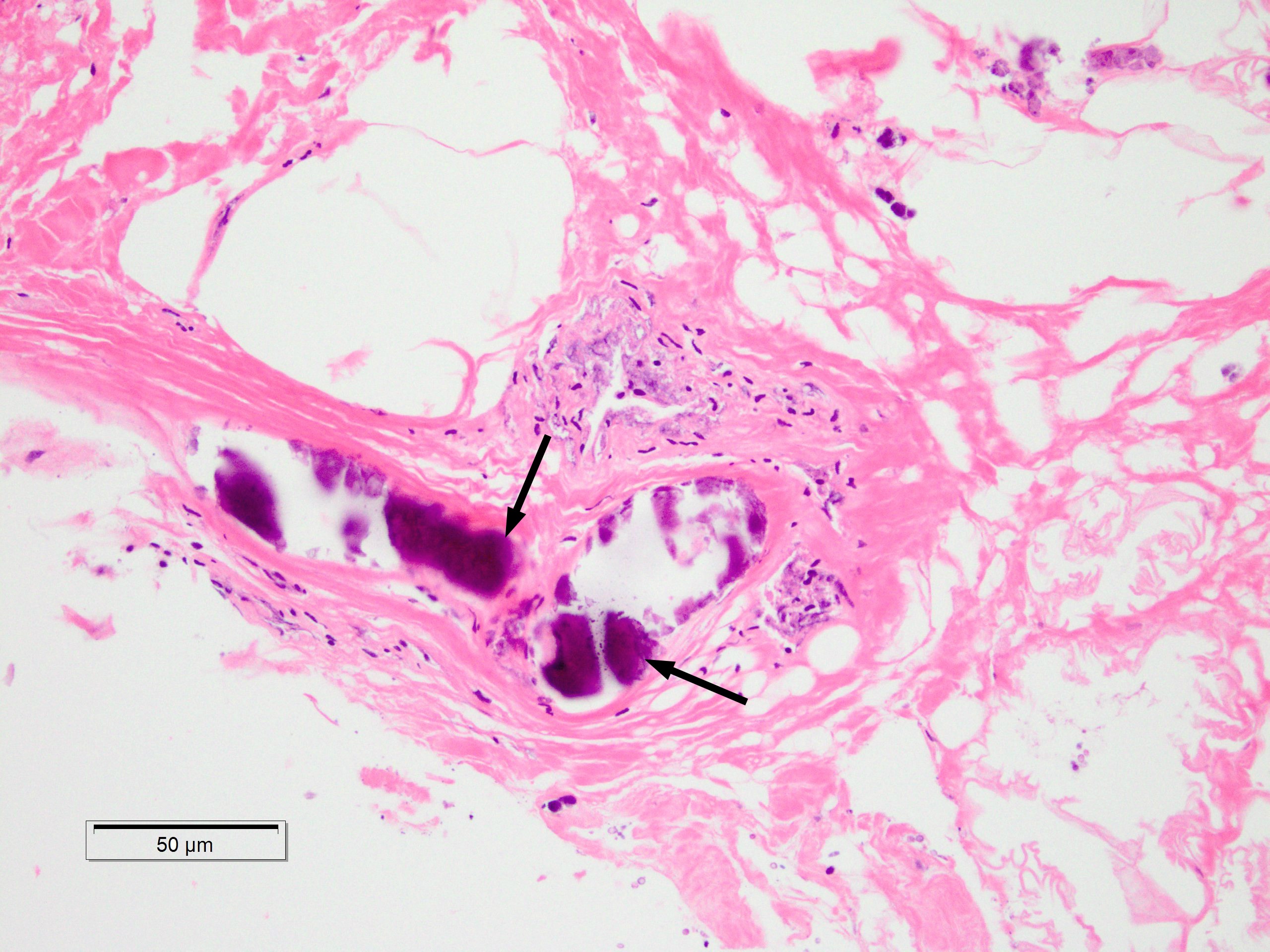
A 43-year old male with a past medical history significant for End Stage Renal Disease (ESRD) (on hemodialysis), tertiary hyperparathyroidism, diabetes mellitus type two, and hypertension presented to UCLA with necrosis of legs, genitalia, and fingers. He had presented to an outside hospital six weeks prior to admission with severe hypotension of unclear etiology. One day thereafter, the patient developed retiform purpura on the medial thighs that evolved into bullae and eventual eschars (see Figure 1-3). The outside hospital differential included vasculitis versus calciphylaxis. Panel of serological autoimmune markers was not done at that time. The patient was treated empirically for vasculitis with several weeks of systemic steroids without improvement of symptoms. No skin biopsy was done. Upon arrival to UCLA, dermatology was consulted on hospital day two, at which point a differential was developed comprised of calciphylaxis, vasculitis, ecthyma gangrenosum, angioinvasive fungi, and coagulopathy. Punch biopsy demonstrated histologic features consistent with calciphylaxis that included deposition of calcium within medium artery intima and media, slight intimal fibrosis, and prominent dermal and epidermal necrosis (see Figure 4). Periodic acid-Schiff and Grocott’s methenamine silver stains x2 were negative for fungal organisms and gram stains x2 were negative for bacterial organisms. Risk factors for calciphylaxis included diabetes with ESRD and associated dysregulation of calcium metabolism. Accordingly, the patient was initiated on sodium thiosulfate 25 grams three times weekly in addition to hyperbaric oxygen therapy. His hospital course was complicated, however, by brief episodes of severe hypotension of unclear etiology and not requiring vasopressors, hypoglycemia, and non-sustained ventricular tachycardia, all of which repeatedly barred the patient from receiving scheduled hyperbaric therapy. Furthermore, following his episodes of hemodynamic instability, the dermatology team consistently noted progression of the patient’s necrotic and purpuric skin lesions, most often on the distal extremities.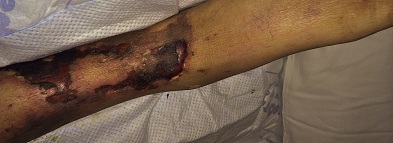
Figure 1: (a) Chemical structures of PAMPS48-PEG227-PAMPS48 (AEA) and PEG47-PMAPTACm (EMm, m = 27,53, and 106).
DISCUSSION
Disordered calcium metabolism is organized into four broad categories: dystrophic, metastatic, idiopathic, and iatrogenic. Calciphylaxis represents the most severe form of metastatic calcification, most commonly occurring in the context of ESRD and secondary hyperparathyroidism, but increasingly reported in the absence of kidney disease in which case it is called Non-Uremic Calciphylaxis (NUC) [8].
Factors that contribute to vascular calcium deposition are reported to include immunosuppression, trauma, infection (particularly human immunodeficiency virus), low protein S levels, metallic salts, certain medications (i.e., albumin infusions, calcium heparinate, warfarin, calcium-based binders, vitamin D analogs, chemotherapy agents, systemic glucocorticoids), increased serum phosphate, higher serum levels of alkaline phosphatase, and treatments for autoimmune conditions such as corticosteroids, methotrexate, and UV light [1,9]. Infectious, autoimmune, and alcoholic hepatitis have also been implicated [1]. The pathophysiology in this case is reported to be mediated by inflammation or acquired thrombophilia from protein C or protein S deficiency [1]. Non-calcium and PTH related etiologies include several conditions rooted in hypercoagulability, which include warfarin use. It has been reported that warfarin leads to a decrease in the endogenous inhibitors of vascular calcification via attenuation of vitamin K-dependent gamma carboxylation of matrix Gla and Gas-6-proteins. Thus, patients treated with warfarin may have a diminished capacity to inhibit vascular calcification due to deceased amounts of active forms of these proteins [1,7]. Primary coagulation disorders implicated include protein S and C deficiency, the antiphospholipid syndrome, vitamin K deficiency, and AT3 deficiency [7].
Other calciphylaxis risk factors include female sex, and obesity or significant weight loss, with the latter two working via effects on fibroelastic septae [2,3,8]. These septae traverse the subcutaneous fat from the deep fascia to the dermis, functioning to anchor the skin to the body, support the adipose tissue, and provide scaffolding for blood vessels that are attached to and incorporated within the septae. In obesity, expansion of the subcutaneous compartment by adipose tissue subjects the septae and associated arterioles to tensile and compressional forces. Phospholipid moieties of damaged cell membranes are exposed and bind calcium to initiate mineralization [9,10]. Any condition that exerts tensile force on the septal-vascular structures may contribute to vascular calcification via a mechanism similar to that outlined for obesity. These conditions include hypoalbuminemic and subsequent edematous states. Bleyer et al., reported a 17-fold increase in the risk of developing calciphylaxis with each decrease in albumin by 1g/L [9]. Other conditions that manifest via a similar mechanism include hypotension or hemodynamic instability [8-11]. Thus, beyond the more obvious decrease in local cutaneous perfusion, hypotension contributes to calciphylaxis progression through contribution to vascular calcification as well.
Although necessary, calcified arterioles alone are insufficient to initiate infarction. Rather, a “second hit” is necessary, consistent with the sensitizer and challenger hypothesis of calciphylaxis pathogenesis put forth by Selye in 1961 [10-16]. At that time, Hans Selye conducted experiments in rats, in which he used agents such as parathyroid extract, high dose vitamin D, high phosphorous diet, or induction of kidney failure for “sensitization” [1]. After a “critical time” period, he introduced the “challenging agent” such as local trauma, albumin, or metallic salts and noted development of cutaneous calcification [1].
There were several key differences between experimental calciphylaxis induced in the Selye rats and calciphylaxis noted in humans. Animals in experimental models did not develop small-artery or arteriolar calcifications. But rather, they developed extensive soft-tissue calcifications [1]. The animals were also able to cast off the calcified molt and replace it with new dermis that did not have features of calciphylaxis [1]. Lastly, experimental calciphylaxis was prevented by administration of glucocorticoids, a fact that contradicts the available data for human calciphylaxis. Nonetheless, Selye and colleagues drew very clear and valid parallels between the “sensitization” and “challenger” model of pathogenesis in experimental rats and humans. In humans, the effect of these “challengers” is magnified with time as the calcified vessels lose the ability to dilate and increase local blood flow in response to hypoperfusion [8]. Additionally, the vascular calcification induces a state of pre-ischemia such that the “second hit” need only be mild [14].
Thus, hypotension and hemodynamic instability contribute in a multifactorial way to calciphylaxis pathogenesis. They directly contribute to vascular calcification via their contribution to septal-vascular tensile stress. They also lead to decreased cutaneous perfusion, which in the presence of pre-existing vascular calcification, may act as the “second hit” necessary to cause cutaneous disease. This multifactorial effect of hemodynamic instability becomes particularly relevant in patients such as ours with end stage renal disease on hemodialysis who may develop post-dialysis hypotension [8].
Limitations of this report include the inherent difficulty to conclude causality. It is possible that progressive calciphylaxis may have contributed to the periods of hypotension and not vice versa. The temporal associations of disease progression with periods of hypotension simply suggest but do not prove causality. Another limitation of the report is the fact that there was no assessment of tissue oxygenation during the periods of hypotension in this patient, which again make it difficult to resolutely prove causality.
CONCLUSION
The exact pathogenesis of calciphylaxis remains unclear and is the topic of continued research and debate. As such, new theories continue to emerge, with a recurrent theme of hypotension speckling the literature [4]. Our case is the first to our knowledge to demonstrate a consistent temporal relationship of episodes of hemodynamic instability with disease progression indicating that aggressive blood pressure control may be a vital component of a multimodal treatment plan in the calciphylaxis patient. The patient referenced in this case deteriorated rapidly. He was transferred to hospice and has since passed away.
REFERENCES
- Nigwekar SU, Kroshinsky D, Nazarian RM, Goverman J, Malhotra R, et al. (2015) Calciphylaxis: risk factors, diagnosis, and treatment. Am J Kidney Dis 66: 133-146.
- Gupta D, Tadros R, Mazumdar A, Moiin A, Fuleihan SF (2013) Breast Lesions with Intractable Pain in End-Stage Renal Disease: Calciphylaxis with Chronic Hypotensive Dermatopathy Related Watershed Breast Lesions. J Palliat Med 16: 551-554.
- Ng AT, Peng DH (2011) Calciphylaxis. Dermatol Ther 24: 256-262.
- Ross EA (2011) Evolution of treatment strategies for calciphylaxis. Am J Nephrol 34: 460-467.
- Wanat KA, Steward CL, Negoianu D, Rosenbach M (2014) Severe Nonuremic Calciphylaxis Due to Hyperphosphatemia Resolving With Multimodality Treatment Including Phosphate Binders. JAMA Dermatol 150: 671-673.
- Amuluru L, High W, Hiatt KM, Ranville J, Shah SV, et al. (2009) Metal deposition in calcific uremic arteriolopathy. J Am Acad Dermatol 61: 73-79.
- Farah M, Crawford RI, Levin A, Chan Yan C (2011) Calciphylaxis in the current era: emerging ‘ironic’ features? Nephrol Dial Transplant 26: 191-195.
- Maroz N, Mohandes S, Field H, Kabakov Z, Simman R (2015) Calciphylaxis in Patients With Preserved Kidney Function. J Am Coll Clin Wound Spec 6: 24-28.
- Mazhar AR, Johnson RJ, Gillen D, Stivelman JC, Ryan MJ, et al. (2001) Risk factors and mortality associated with calciphylaxis in end-stage renal disease. Kidney Int 60: 324-332.
- Janigan DT, Hirsch DJ (2006) Does obesity play a role in the pathogenesis of calcific uraemic arteriolopathy? Nephrol Dial Transplant 21: 865-868.
- Janigan DT, Hirsch DJ, Klassen GA, MacDonald AS (2000) Calcified subcutaneous arterioles with infarcts of the subcutis and skin (“calciphylaxis”) in chronic renal failure. Am J Kidney Dis 35: 588-597.
- Verdalles U, Cueva Pde L, Verde E, Vinuesa SG, Goicoechea M et al. (2008) Calciphylaxis: A Severe Complication of the Cardiometabolic Syndrome in Patients Receiving Hemodialysis. J Cardiometab Syndr 3: 63-67.
- Selye H (1962) Calciphylaxis. Chicago, IL, University of Chicago Press, USA.
- Grob JJ, Legre R, Bertocchio P, Payan MJ, Andrac L, et al. (1989) Calcifying panniculitis and kidney failure. Considerations on pathogenesis and treatment of calciphylaxis. Int J Dermatol 28: 129-131.
- Janigan DT, Perey B, Marrie TJ, Chiasson PM, Hirsch D (1997) Skin necrosis: an unusual complication of hyperphosphatemia during total parenteral nutrition therapy. JPEN J Parenter Enteral Nutr 21: 50-52.
- Janigan DT, Prokopetz RD, Chawla S, Durning RG (1989) Massive necrosis of fat and skin as complication of obesity. CMAJ 140: 665-668.
Citation: Higgins S, Hu M, Worswick S, Binder S (2016) Calciphylaxis and Hypotension: Report of a Case and Review of the Literature. J Clin Dermatol Ther 3: 019.
Copyright: © 2016 Shauna Higgins, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.