Case of MOG Seropositive longitudinally extensive Transverse Myelitis
*Corresponding Author(s):
Amina SaddiqaResident Neurology, Department Of Neurology, Pakistan Institute Of Medical Sciences, Islamabad, Pakistan
Tel:03361506877,
Email:aminasaddiqa@gmail.com
Abstract
Background: Previously, anti-MOG-associated disorders were thought to involve seronegative CNS disorders. However, relapse patterns and prognoses of these disorders are distinctive enough that accurate diagnoses may be made. Immunotherapeutic agents, although not all, appear to successfully alleviate the disease. Therefore, more case reports are needed to broaden the clinical spectrum of disease along with larger cohort studies. Only a few cases were reported from Pakistan.
Case: Most patients with MOG-associated disorders exhibit a similar clinical spectrum as that of Multiple Sclerosis and NMOSD; here, we present a case of a young male with subacute onset paraplegia with sphincters involvement and patchy loss of pinprick sensation with positive Anti-MOG Antibody.
Keywords
LETM; MOG; MS; NMOSD;
Introduction
Myelin oligodendrocyte glycoprotein is located on the outermost layer of the myelin sheath which makes it particularly susceptible to immune-mediated demyelination. Myelin Oligodendrocyte Glycoprotein Antibody-associated Disease (MOGAD) is an inflammatory disease of the Central Nervous System (CNS) characterized by attacks of immune-mediated demyelination. Its clinical course can be monophasic and relapsing, with clinical presentation resembling Acute Disseminated Encephalomyelitis (ADEM), Optic Neuritis (ON), transverse myelitis, and brainstem demyelinating syndrome. MRI and electrophysiological testing like VEP findings should be compatible with CNS demyelination, and there should be seropositivity of MOG antibody serology detected by cellular assay (which is sensitive and specific) to establish its diagnosis [1]. MOG-Ab-associated acute transverse myelitis is a relatively common presentation of MOGAD in adults and can be seen in children as well. Diagnostic criteria of MOGAD required presence of core clinical demyelinating event which includes optic neuritis , transverse myelitis , Acute Disseminated Encephalomyelitis , cerebral monofocal or polyfocal deficits , brainstem or cerebellar deficits , cerebral cortical encephalitis , often with seizures [2], positive MOG antibody and exclusion of other diagnosis.
Case Presentation
A 27 years old man presents to the emergency department with 7 days history of bilateral lower limb weakness associated with urinary retention and constipation. Lower limb weakness was sudden in onset, symmetrical and rapidly progressive. He had no history of numbness, paresthesia and backache. Upper limbs were normal. There was no history of visual impairment, dysarthria, dysphagia, no history of nasal regurgitation, no breathing difficulty. He had no medical history, in particular, no history of TB contact. Family history is not significant. He is married, a waiter by profession, issue-less, and belongs to a middle income family. He is a non-smoker, not taking any medications regularly.
In his physical examination, he was of average height and built, conscious, and oriented in time, place, and person. There was no visible cyanosis, clubbing, pallor, jaundice, lymphadenopathy, thyroid enlargement, and edema. His oral temperature is 98.6°F (37°C), pulse was regular and 94 beats/minute, blood pressure was 110/70 mm Hg, respiratory rate was 16 breaths/minute with oxygen saturation of 98% at room air. There were no signs of meningeal irritation. His upper limbs had normal tone with MRC grade 5 power, and his deep tendon reflexes were intact. In lower limbs, he had 0/5 power in proximal and distal muscles bilaterally according to MRC grading, with increased tone and grade 3 deep tendon reflexes. His plantar reflex were bilaterally extensor. Cerebellar functions were intact in both upper limbs. In lower limbs, cerebellar functions could not be assessed due to MRC grade zero power. His gait could not be assessed. On sensory system examination, there was patchy impairment of sensations in thighs and some parts of calves but no definite sensory level. Joint position sensation was intact. No gross spinal deformity and no spinal tenderness.
On eye examination, his pupils were bilaterally equal and reactive to light, with intact extraocular movements. His visual acuity in the right eye is 6/18 and in the left eye 6/18, but no relative afferent pupillary defect, colour vision impairment or disc pallor . No relative afferent pupillary defect. The rest of the cranial nerves examination was unremarkable. His cardiovascular, respiratory and gastrointestinal examination was unremarkable.
His laboratory analysis demonstrates normal complete blood count, renal and liver function tests, serum electrolytes, coagulation profile, and urine routine examination. His hepatitis B and C screening is negative.cerebrospinal fluid analysis was done.
|
CSF |
R/E |
|
Color |
Light red |
|
Turbidity |
Slightly turbid |
|
WBCs |
90/mm3 |
|
xanthochromia |
Absent |
|
Neutrophils |
9% |
|
Lymphocytes |
91% |
|
Protein |
85.4mg/dl |
|
Glucose |
51mg/dl |
On neuroimaging, MRI cervico-thoracic spine showed T2 hyperintense lesion extending from the lower cervical to the mid-thoracic cord with minimal cord expansion, with no post-contrast enhancement, likely due to inflammatory demyelinating aetiology. (Figure 1 & 2) MRI Brain with contrast done in this patient, was reported as an essentially unremarkable study with no evidence of demyelinating plaques or any other significant abnormality (Figure 3).
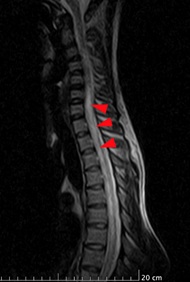 Figure 1: T2 sagittal view hyperintense signal in lower cervical and midthoracic region.
Figure 1: T2 sagittal view hyperintense signal in lower cervical and midthoracic region.
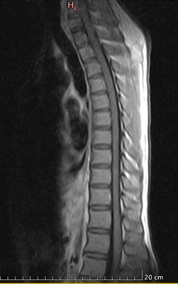 Figure 2: T1 with contrast sagittal view shows no post contrast enhancement.
Figure 2: T1 with contrast sagittal view shows no post contrast enhancement.
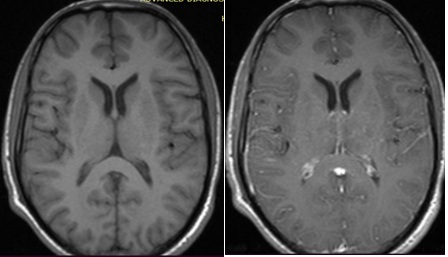 Figure 3: MRI Brain Axial view, plain (left) and with contrast (right) showed no contrast enhancing lesion reported as unremarkable study.
Figure 3: MRI Brain Axial view, plain (left) and with contrast (right) showed no contrast enhancing lesion reported as unremarkable study.
VEP done which was reported as normal.
His HIV serology and VDRL are negative. NMO (aquaporin-4 IgG) is also negative. The anti-MOG antibody test is positive in this patient. Pulse therapy was initiated, but no real improvement in his condition. Plasmapheresis was initiated and 4 sessions of Plasmapheresis were done and he showed marked improvement in power to 3/5 in both lower limbs. General supportive care started and counselled in detail regarding physiotherapy and called for follow-up visits.
Discussion
Myelin oligodendrocyte glycoprotein (MOG) is a protein on the surface of oligodendrocytes, is highly immunogenic and its exact function is unknown; it may act as a cell adhesion molecule, and its location on the outermost part of myelin sheath makes it more susceptible to immune-mediated CNS demyelination. It is a minor component of myelin sheath comprising 0.5% [3]. It was previously considered a potential antibody target for Multiple sclerosis patients, but since 2007 MOG-associated Immunoglobulin (IgG) is considered a separate demyelinating disease. It is a rare autoimmune disorder [4]. MOG-IgG-associated disorders present mostly as Acute Disseminated Encephalomyelitis (ADEM) in children, optic neuritis, and severe myelitis in adults. Currently, few studies showed that it can present as brainstem and autoimmune encephalitis [5]. MOG-associated antibody disease showed a female predominance as compared to males [6]. Previously, the most common clinical and radiological presentation in MOG antibody-associated disease was optic neuritis [6] but in our case, the patient’s presenting complaint was bilateral lower limb weakness, and we found decreased visual acuity in this case as an incidental finding.
Transverse myelitis is an initial presentation mainly in adults who are seropositive for MOG-AD. It is a severe, disabling condition, and with the help of antibody assessment for NMOSD and MOG, we are able to identify the causes of transverse myelitis. MOG-IgG-associated myelitis presents as acute flaccid myelitis. MOG-AD transverse myelitis is mostly longitudinally extensive (LETM). As in our case, MRI dorsolumbar spine showed a longitudinally extensive lesion extending from the lower cervical to the mid-thoracic region. It showed no post-contrast enhancement as compared to AQP4-IgG seropositive myelitis. Deep grey matter lesions are more common in MOG-IgG myelitis [7]. It can also be associated with optic neuritis and brainstem encephalitis. We can differentiate MOG-AD from NMOSD and MS, although there are few clinical and radiological similarities between MOG-AD and NMOSD but can easily differentiate from MS. Silent lesions which are commonly found in MS are absent in MOGAD [1]. MOGAD has the greatest predilection for conus medullaris resulting in sphincter involvement and bladder dysfunction [8].
MOG-AD seropositive patients showed better response to steroids pulse therapy as compared to both NMOSD and MS patients. Long-term clinical outcomes in MOG-AD showed mostly partial or nearly complete recovery [8]. Residual cognitive deficits remained in children mainly and very few patients among adults showed poor functional outcomes in the long run. Prognosis is generally favorable in MOG seropositive patients but severe residual disability can occur if the diagnosis is missed due to the nonavailability of antibody testing. Early diagnosis helps us in differentiating the cause of longitudinally extensive transverse myelitis and prevent residual attacks, for a long-term management plan.
Declaration
- Ethical approval
It has been approved by Departmental Review Board and Ethics Committee, Neurology Department of Pakistan Institute Of Medical Sciences.
- Consent to participate and publication
Consent to participate and publication has been taken by participant.
- Competing interest
The authors declare that they have no competing interests.
Funding
No funding sources.
Author’s contributions
All authors participated in data collection, manuscript writing .All authors read and approved manuscript.
Acknowledgements
I am really grateful to my co-authors who contributed in this study.
References
- Jurynczyk M, Jacob A, Fujihara K, Palace J (2019) Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease: Practical considerations. Pract Neurol 19: 187-195.
- Banwell B, Bennett JL, Marignier R, Kim HJ, Brilot F, et al. (2023) Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International Mogad Panel proposed criteria. Lancet Neurol 22: 268-282.
- Dos Passos GR, Oliveira LM, da Costa BK, Apostolos-Pereira SL, Callegaro D, et al. (2018) Mog-IGG-associated optic neuritis, encephalitis, and myelitis: Lessons learned from Neuromyelitis Optica Spectrum disorder. Front Neurol 9: 217.
- Salama S, Khan M, Pardo S, Izbudak I, Levy M (2019) Mog antibody-associated encephalomyelitis/encephalitis. Mult Scler 25: 1427-1433.
- Hegen H, Reindl M (2020) Recent developments in Mog-IGG Associated Neurological Disorders. Ther Adv Neurol Disord 13.
- Paige Sutton, Lutz MW, Hartsell FL, Kimbrough D, Tagg NT, et al. (2022) Myelin oligodendrocyte glycoprotein (MOG) antibody-associated disease: Presentation and outcomes of adults at a single center. J Neuroimmunol 373:577987.
- Dubey D, Pittock SJ, Krecke KN, Morris PP, Sechi E, et al. (2019) Clinical, radiologic, and prognostic features of myelitis associated with myelin oligodendrocyte glycoprotein autoantibody, JAMA Neurol 76: 301-309.
- Sechi E, Cacciaguerra L, Chen JJ, Mariotto S, Fadda G, et al. (2022) Myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD): A review of clinical and MRI features, diagnosis, and Management. Front Neurol 13:885218.
Citation: Saddiqa A, Rajput HM, Khalil M, Farooq MA, Waqar Z, et al. (2023) Case of MOG Seropositive longitudinally extensive Transverse Myelitis. J Brain Neursci 7: 025.
Copyright: © 2023 Amina Saddiqa, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.