Characteristics of the Phenotype-Genotype Correlation in Cohort of Russian and Ukrainian Patients with Lysosomal Acid Lipase Deficiency
*Corresponding Author(s):
Kamenets ElenaFsbsi Research Centre For Medical Genetics, Moskvorechie 1, 115522 Moscow, Russian Federation
Email:elenakamenec@yandex.ru
Abstract
Lysosomal Acid Lipase Deficiency (LAL-D) is a rare autosomal recessive disease caused by mutations in the LIPA gene encoding the enzyme of cholesterol metabolism Lysosomal Acid Lipase (LAL). The common variant of LIPA is c.894G>A caused the disease in more than ? half of cases in Europe.
The aim of our work was to study LIPA gene mutations spectrum and find correlations of the genotype and secondary biochemical markers in cohort of Russian and Ukrainian LAL-D patients.
In 39 patients LAL-D diagnosis was confirmed. Molecular analysis of LIPA revealed the variant c.894G>A in homozygous or compound-heterozygous state in 34 of them. Fifteen other variants were defined, 11 of them were novel.
Chitotriosidase activity and the oxysterols levels of homozygous and compound-heterozygous for c.894G>A patients were compared.
Two common mutations among the Russian and the Ukrainian patients were defined. Seventeen identified variants with 11 of them novel and unique demonstrate the variety of LIPA mutations spectrum in Russia.
Heterozygous sever defects of LIPA were found to increase the chitotriosidase activity and the oxysterols levels in the patients. It indicates the higher extent of macrophages activation/liver fibrosis and dyslipidemia progression, respectively. These data may have a prognostic significance.
Keywords
Chitotriosidase; Cholesterol; Lysosomal acid lipase deficiency; LIPA gene; Lysosomal storage diseases; Oxysterols
ABBREVIATIONS
7-KC - 7-Ketocholesterol Concentration
ALT - Alanine Aminotransferase
AST - Aspartate Aminotransferase
CESD - Cholesteryl Ester Storage Disease
Cht - Chitotriosidase
CT - Computer Tomography
C-triol - Cholestane-3β,5α,6β-triol
DBS - Dried Blood Spots
HDL - High Density Lipoprotein
LAL - Lisosomal Acid Lipase
LAL-D - Lisosomal Acid Lipase Deficiency
LDL - Low Density Lipoprotein
LOF - mutation - mutation caused total Loss of Enzyme Function
LSD - Lysosomal Storage Disease
TC - Total Cholesterol
TG - Triglycerides
US - Ultrasound
WD - Wolman Disease
INTRODUCTION
Lysosomal Acid Lipase Deficiency (LAL-D; MIM#278000) is a rare autosomal recessive Lysosomal Storage Disease (LSD) caused by mutations in the LIPA (MIM 613497) gene encoding the enzyme Acid Lipase (LAL) that plays a key role in lipid metabolism. LAL splits LDL-derived neutral lipids into free cholesterol and fatty acids. Dysfunction of the enzyme leads to progressive accumulation of Cholesteryl Esters (CE) and Triglycerides (TG), mainly in cells of the macrophage/monocyte system of the liver and spleen and hepatocytes, too [1,2]. LAL-D results in the impairment of liver function in the form of elevation of serum aminotransferases and dyslipidemia.
LAL-D is considered as a continuum of phenotypes, historically divided into two clinical forms: Wolman Disease (WD) with neonatal onset and rapidly progressing fatal course, and Cholesteryl Ester Storage Disease (CESD) with late onset, mild course and a varied severity of symptoms [2].
LIPA is located on the chromosome 10q23.31. Over 60 variants of this gene are known to be associated with LAL-D. The c.894G>A (E8SJM-1G>A) variant is the most common identified in European populations and is responsible for more than half of reported cases [2-5]. For some other alleles, an ethnic background is observed [3,6]. Patients harboring the common c.894G>A allele or a missense mutation that ensures the significant residual enzyme activity in at least a heterozygous state, have CESD phenotype, regardless how severe is the other variant. WD phenotype is the result of two severe deleterious alleles in a homozygous or compound-heterozygous state [2,3,7].
The definite incidence of LAL-D is unknown. The disease is considered as pan-ethnic and its frequency in Europe is estimated from 1/300 000 to 1/40 000 depending on ethnicity and geographical location [2,7-9].
The measurement of LAL activity could be carried out in cultures of skin fibroblasts, peripheral leukocytes or liver tissue cells. The method used for the measurement in Dried Blood Spots (DBS) in the presence of specific inhibitor (Lalistat-2) introduced by Hamilton et al., in 2012 has been found to be effective and the most rapid; nowadays it is considered as the first diagnostic step for LAL-D screening [10].
There are also present other secondary biochemical markers of the disease. Chitotriosidase (Cht) is a human chitinase secreted by the activated macrophages. Cht activity is a classic marker of gaucher disease and moderately elevated values are found in a variety of some other LSDs including LAL-D [11]. Due to the storage process and subsequent macrophages activation, Cht activity is increased in the course of the disease, so it could be considered as a serum marker for LAL-D. Oxysterols are oxidized derivatives of cholesterol, almost always produced by non-enzymatic processes. Two such compounds, cholestane-3β,5α,6β-triol (C-triol) and 7-Ketocholesterol (7-KC), have been shown to be elevated in patients with Niemann-Pick type C disease, cerebrotendinous xanthomatosis and also LAL-D [12].
The aim of our work was to study LIPA gene mutations spectrum and find correlations of the genotype and secondary biochemical markers, including serum chitotriosidase activity, cholestane-3β,5α,6β-triol and 7-ketocholesterol levels, in the cohort of Russian and Ukrainian LAL-D patients.
MATERIALS AND METHODS
Patients
Between 2009 and 2015, 11 patients (5 male and 6 female) from 10 unrelated families, with age ranged from 2 month to 23 years old, suspected to have LAL-D based on clinical and biochemical features, were referred for molecular analysis of the LIPA gene.
Since 2016 the enzymatic assay for LAL activity in DBS in the presence of Lalistat-2 was added to the list of diagnostic screening tests on LAL-D carried out in Research Centre for Medical Genetics of Moscow.
The selective criteria (uncombined) of suspected LAL-D were the following:
- Hepatomegaly, feeding difficulties, frequent vomiting, diarrhea, swelling of the abdomen and anemia presented in the first 6 months of life
- Hepatomegaly observed in childhood
- ALT > 1,5-fold upper limit of normal
- Presence of dyslipidemia type II
From January 2016 till January 2019, a total number of tested DBS specimens were 1998. Patients were originated from medical centers of different regions from Russia and Ukraine. Sex distribution was approximately equal, the age varied from the first week of life to 45 years.
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients included in the study or from their legal guardians.
Patients’ medical records
A retrospective chart review of patients’ medical records concerning the first presented signs and symptoms, as well as biochemical (liver transaminases, platelet count, total cholesterol, HDL-C, LDL-C, TG), histological (histopathological examination of liver biopsy specimens) were collected. Some clinical and laboratory data have been published previously [13,14]. An informed consent for collecting clinical data and blood samples for biobanking was obtained from all patients included in the study.
DNA sequencing
DNA was extracted from whole blood with EDTA and DBS using the DNAPrep100 Kit (IsoGene, Moscow, Russia).
Coding exons of LIPA (exons 2 - 10) were amplified by PCR and analyzed by direct sequencing on ABI PRISM 3500xL Genetic Analyzer (Applied Biosystems, USA). Primers sequences and PCR protocols are available on request. Analysis of Sanger sequencing data was performed using Chromas software and Nucleotide-BLAST software. LIPA variants are numbered according to the guidelines from the Human Genome Variation Society (http://www.hgvs.org/mutnomen) using NCBI Reference Sequences NM_000235.3, NG_008194.1.
Biochemical assays
LAL activity was measured by fluorimetric analysis in DBS as was reported by Hamilton et al. [10]. Cht activity was measured by fluorimetric analysis using the method described by vom Dahl et al., and adapted for DBS, according to own adaptation (unpublished) [11]. Oxysterols (CT and 7-KC) levels were measured by HPLC-MS/MS as described by Boenzi et al., with modification [15]. Chromatographic separation was performed on column Gold C18 (2.1X100 mm, 5 microns) using HPLC-system LC20 (Shimadzu, Japan) with detection on mass-spectrometer Sciex 3200Trap (ABSciex, USA).
Determination of variant pathogenicity
Classification of novel variants by their pathogenicity was performed according to standards and guidelines for the interpretation of sequence variants [16]. For missense and intronic variants, was based on minor allele frequencies of the mutation detected by genetic sequencing (GnomAD, PMID: 27535533), parental segregation analysis (if performed) and in silico pathogenicity prediction algorithms. In silico analysis was performed using predictive algorithms/software MutationTaster, PolyPhen2, SIFT, PROVEAN (for missense variants), Human Splicing Finder and NetGene2 (for variants of alternative splicing). For novel synonymous mutation c.600G>A, determination of pathogenicity was based on cDNA analysis by direct sequencing and functional analysis by minigene splicing assay.
Statistic analysis
Statistic analysis was performed using GraphPad Prism software (version 6.01).
RESULTS AND DISCUSSION
Thirty-nine patients with LAL-D were enrolled into the study. The ethnic distribution was as follows: 32 Russians, 6 Ukrainians, 1 Azerbaijani. The diagnosis was confirmed by molecular analysis of the LIPA gene or by the measurement of LAL activity in DBS and molecular analysis. The LAL activity in affected patients was noted as less than 0.04 nmol/punch/h.
Genetic analysis
Among 39 patients, 34 had a common splice-junction c.894G>A variant of LIPA, identified in homozygous (15 pts) or compound-heterozygous (19 pts) state. Eight patients had c.796G>T (p.G266*) variant identified only in compound-heterozygous state. We also found the 4 other known mutant alleles: c.309C>A, c.347G>A, c.398delC, c.894+1G>A and 11 novel variants. The latter were presented in Azerbaijani and some of Russian patients. Some of the novel variants were published by us earlier without any pathogenicity analysis [17].
Three out of eleven novel variants were classified as nonsense substitutions: c.348G>A (p.W116*), c.398C>A (p.S133*) and c.420G>A (p.W140*), what is “very strong” criterion of pathogenicity [16]. The other five variants were classified as leading to frame-shift alterations: 167_170dup4 (p.L61Yfs*5), c.421delG (p.A141Lfs*20), c.442delG (p.A148Qfs*13), c.817_818delAA (p.N273Yfs*3), c.911_912delinsT (p.K304Ifs*4) and the substitution c.-3A>G (p.?) that affects the canonical acceptor splicing site. In addition, there are the described mutations in codons 116, 133 and 140 leading to the same predictable changes in the primary structure of the enzyme (“strong” criterion of pathogenicity [16]). The bioinformatic analysis of the substitution c.-3A>G with Human Splicing Finder and NetGene-2 showed a high probability of alteration of the constitutive acceptor splicing site in intron 1, that resulted in skipping of exon 2 and the initiating codon of LIPA gene cDNA. The synonymous variant c.600G>A in exon 6 showed a high probability of exonic cryptic splicing site activation leading to 63 b.p. deletion corresponding to c.541_603del63 of the cDNA. The variant c.600G>A was inherited from both patient’s parents, who were heterozygous, and the variants c.-3A>G, c.398C>A and c.420G>A reside in trans-configuration with the variant c.894G>A, subsequent to the results of the parental segregation analysis (“moderate” criterion of pathogenicity [16]). For the variant c.600G>A the functional analyses by minigene assay was performed [14].
The remaining variant is the missense substitution c.956A>T (p.H319L). Predictable pathogenicity of these missense variant was valued as possible damaging.
In accordance to the gnomAD database, the allelic frequencies of all defined variants in the healthy cohort were extremely low (Table 1). A retrospective analysis of the exomic sequencing results of 50 DNA samples was performed (100 chromosomes). The samples were provided by patients who applied to the Research Centre for Medical Genetics of Moscow with different diagnoses and had LAL activity in DBS at the reference range. There were not any of revealed in LAL-D patients variants in the 100 chromosomes of patients with normal LAL activity.
|
? |
Variant |
Effect |
Allelic frequency (gnomAD) |
HGMD ID |
References |
PolyPhen-2 |
SIFT |
PROVEAN |
Variant significance |
|
Known variants |
|||||||||
|
1 |
c.309C>A (p.S103R) |
Missense |
0.000008 |
CM1617783 |
[5,18] |
Probably damaging |
Damaging |
Deleterious |
Likelypathogenic |
|
2 |
c.347G>A (p.W116*) |
Nonsence |
ND |
CM993350 |
[19-21] |
- |
- |
- |
Pathogenic |
|
3 |
c.398delC (p.S133*) |
Nonsence |
0.00002 |
HD971471 |
[3,22] |
- |
- |
- |
Likelypathogenic |
|
4 |
c.796G>T (p.G266*) |
Nonsence |
ND |
CM960945 |
[19,20,23] |
- |
- |
- |
Pathogenic |
|
5 |
c.894G>A(p.275_298del) |
Synonymous |
0.000827 |
CS951467 |
[4,21] |
High probability of donor splice site alteration (analyzed by Human Splicing Finder and NetGene-2) |
Pathogenic |
||
|
6 |
c.894+1G>A (p.275_298del) |
Spliceregion alteration |
ND |
CS963024 |
[19,23] |
High probability of donor splice site alteration (analyzed by Human Splicing Finder and NetGene-2) |
Pathogenic |
||
|
Novel variants |
|||||||||
|
1 |
c.-3A>G (p.?) |
Spliceregion alteration |
ND |
- |
- |
- |
- |
- |
Likelypathogenic |
|
2 |
c.177_180dup4 (p.L61Yfs5*) |
Frame-shift |
ND |
- |
- |
- |
- |
- |
Likelypathogenic |
|
3 |
c.348G>A (p.W116*) |
Nonsence |
ND |
CM172457 |
[17] |
- |
- |
- |
Pathogenic |
|
4 |
c.398C>A (p.S133*) |
Nonsence |
ND |
CM1724568 |
[17] |
- |
- |
- |
Pathogenic |
|
5 |
c.420G>A (p.W140*) |
Nonsence |
ND |
CM1724569 |
[17] |
- |
- |
- |
Pathogenic |
|
6 |
c.421delG (p.A141Lfs*20) |
Frame-shift |
ND |
- |
- |
- |
- |
- |
Likelypathogenic |
|
7 |
c.442delG (p.A148Qfs*13) |
Frame-shift |
ND |
- |
- |
- |
- |
- |
Likelypathogenic |
|
8 |
c.600G>A (p.180_200del) |
Cryptogenic splice site |
ND |
- |
- |
High probability of exonic cryptic splice site activation (analyzed by Human Splicing Finder and NetGene-2); 63 b.p. deletion in mRNA |
Pathogenic |
||
|
9 |
c.817_818delAA (p.N273Yfs3*) |
Frame-shift |
ND |
CD1724570 |
[17] |
- |
- |
- |
Likelypathogenic |
|
10 |
c.911_912delinsT (p.K304Ifs4*) |
Frame-shift |
ND |
- |
- |
- |
- |
- |
Likelypathogenic |
|
11 |
c.956A>T (p.H319L) |
Missense |
ND |
- |
- |
Possibly damaging |
Damaging |
Deleterious |
Uncertain significance |
Table 1: The classification of defined LIPA variants.
Taking all combined data into account, the variants were classified by their functional and clinical significance according to standards and guidelines for the interpretation of sequence variants and presented in table 1 [16].
In our cohort, like in other reported, the common mutation c.894G>A in a homozygous or compound-heterozygous state also predominates. It’s allelic frequency amounts 62.8%, what corresponds with reported data about European populations. A high frequency of this variant in Europe and Russia could be explained by the founder effect and confirmed by some studies of its’ co-segregated with a unique haplotype [3]. The same suggestion is applicable for c.796G>T variant in Russian and Ukraine patients (8 alleles, 10.3%).
Genotype/phenotype correlations
Patients with Wolman disease
Three of diagnosed patients presented with WD. Two of them were compound-heterozygous for one known pathogenic variant, c.398delC and c.796G>T, respectively and for the new variant c.348G>A. The third patient was compound-heterozygous for two new variants: c.442delG and c.817_818delAA.
All five variants are so-called LOF-mutations (“loss of function”), leading to a complete loss of the activity of the enzyme [16].
In all 3 children the disease manifested in the first months of life. The first symptoms were severe hepatosplenomegaly and anemia, one patient was also hypotrophic. In the bone marrow smears, a delayed leukocyte maturation and accumulation of macrophages with “foamy” cytoplasm were found. US and CT scans of the abdomen in all three patients revealed hepatosplenomegaly and calcifications in the adrenal glands. Increased levels of liver transaminases, serum bilirubin and total as well LDL cholesterol were observed. The main signs and symptoms and biochemical parameters of the patients are presented in table 2. The overall condition of patients rapidly deteriorated due to multiple organ failure. They finally succumbed 1-2 months after initial symptoms.
|
? |
Sex |
First signs and symptoms
|
Age of diagnosis |
First consultation– laboratory data |
Liver biopsy (if performed); Liver in US or CT examination (if performed) |
Activity of the enzymes |
Oxy-sterols levels |
Genotype |
|
Homozygous for the variant c.894G>A patients |
||||||||
|
1 |
M |
recurrent abdominal pain, vomiting;
hepatomegaly (4y) |
14y |
ALT 180.0 AST 95.0 TC 8.35 HDL n.d. LDL n.d. TG 0.94 |
Decreased echogenicity of liver in US, hyperechogenic inclusions in the parenchyma (calcifications); |
LAL 0.00
Cht752 |
No data |
c.894G>A/ c.894G>A |
|
2 |
M |
enlarged abdomen;
hepatosplenomegaly (3y) |
5y |
ALT 104.0 AST 89.4 TC 9.68 HDL 0.58 LDL 7.95 TG n.d. |
Hepatosplenomegaly and lymphadenopathy in CT;
Liver biopsy – lipid storage vacuoles in hepatocytes and Kupffer cells |
LAL 0.00
Cht 25
|
C-triol 24.4 7-KC 99 |
c.894G>A/ c.894G>A |
|
3 |
F |
recurrent abdominal pain;
elevated transaminases (7y) |
8y |
ALT 163.0 AST 131.0 TC 9.33 HDL 1.20 LDL 7.53 TG 1.69 |
Diffuse heterogeneity, increased echogenicity of liver in US; |
LAL 0.00
Cht 61
|
C-triol 48.9
7-KC 223 |
c.894G>A/ c.894G>A |
|
4 |
F |
no complain;
elevated transaminases (6y) |
7y |
ALT 124.0 AST 81.0 TC 9.55 HDL 0.92 LDL 8.40 TG 1.34 |
Diffuse heterogeneity and increased echogenicity of liver in US;
|
LAL 0.01
Cht58
|
C-triol33.4
7-KC 189.6 |
c.894G>A/ c.894G>A |
|
5 |
F |
recurrent abdominal pain;
elevated transaminases, hyperbilirubinemia (2y) |
9 y |
ALT 183.2 AST 144.0 TC 6.16 HDL 1.28 LDL 4.60 TG 0.94 |
Diffuse heterogeneity, increased echogenicity of liver in US;
|
LAL 0.00
Cht 39 |
No data |
c.894G>A/ c.894G>A |
|
6 |
F |
recurrent icteric sclerae, apathy;
mild hepatomegaly, elevated transaminases (9y) |
19y |
ALT 70.0 AST 54.0 TC 7.80 HDL 1.53 LDL 5.32 TG n.d. |
Diffuse changes of liver parenchyma, signs of fibrosis in US;
Liver biopsy (10y) – periportal fibrosis |
LAL 0.01
Cht 334 |
No data |
c.894G>A/ c.894G>A |
|
7 |
F |
no complain;
elevated transaminases (5y) |
13y |
ALT 114.0 AST 148.0 TC 8.20 HDL 1.10 LDL 6.19 TG 1.97 |
Diffuse changes of liver parenchyma in US; Liver biopsy – periportal fibrosis Liver fibrosis in METAVIR score F-3 |
LAL 0.00
Cht 15 |
No data |
c.894G>A/ c.894G>A |
|
8 |
M |
enlargedabdomen;
hepatomegaly (7m) |
32y |
ALT 188.0 AST 80.0 TC 7.01 HDL 0.80 LDL 5.02 TG 2.56 |
Features of liver fibrosis in US |
LAL 0.00
Cht 32 |
C-triol42.3
7-KC 104.4 |
c.894G>A/ c.894G>A |
|
9 |
F |
recurrent abdominal pain;
hepatosplenomegaly, elevated transaminases (5y) |
7y |
ALT 134.0 AST 86.0 TC 8.00 HDL 1.00 LDL 7.07 TG n.d. |
Diffuse heterogeneity of liver parenchyma in US |
LAL 0.00
|
No data |
c.894G>A/ c.894G>A |
|
10 |
M |
recurrent abdominal pain, poor appetite, vomiting;
elevated transaminases (4y) |
6y |
ALT 88 AST 125 TC 5.90 HDL 1.05 LDL 4.58 TG 0.94 |
Mildly increased echogenicity of liver in US;
Liver biopsy (6y) – fibrosis, lipid storage vacuoles in hepatocyes and Kupffer cells; Liver fibrosis in METAVIR score F2-F3 |
LAL 0.03
Cht 42 |
C-triol26.7
7-KC 137.5 |
c.894G>A/ c.894G>A |
|
11 |
F |
no complain;
hepatomegaly, elevated transaminases (3y) |
9y |
ALT 210.0 AST 146.0 TC 11.10 HDL 1.14 LDL 9.08 TG 1.51 |
Diffuse heterogeneity, increased echogenicity of liver in US;
|
LAL 0.02
Cht 220 |
C-triol101.1
7-KC 181.8 |
c.894G>A/ c.894G>A |
|
12 |
F |
no complain;
mild hepatomegaly, elevated transaminases (2y) |
5y |
ALT 160.0 AST 118.0 TC 9.07 HDL 1.45 LDL 8.24 TG n.d. |
Increased echogenicity of liver in US;
Liver biopsy (3y) – lipid storage vacuoles in hepatocytes and Kupffer cells |
LAL 0.02
|
No data |
c.894G>A/ c.894G>A |
|
13 |
F |
no complain;
mild hepatomegaly (1y 6m) |
2y |
ALT 32.0 AST 58.0 TC 6.41 HDL 0.60 LDL 3.79 TG 1.68 |
Normal echogenicity of liver in US |
LAL 0.03
Cht 54 |
C-triol21.3
7-KC 144.3 |
c.894G>A/ c.894G>A |
|
14 |
M |
recurrent vomiting, jaundice;
hepatomegaly, elevated transaminases (4y) |
6y |
ALT 117.0 AST 97.0 TC 7.89 HDL 0.84 LDL 6.93 TG n.d. |
Diffuse heterogeneity, decreased echogenicity of liver in US
|
LAL 0.00
Cht 884 |
No data |
c.894G>A/ c.894G>A |
|
15 |
F |
no complain;
elevated transaminases, dyslipidemia (15y) |
17y |
ALT 114.0 AST 89.0 TC 11.2 HDL 1.55 LDL 9.63 TG n.d. |
Increased echogenicity of liver in US; |
LAL 0.01
Cht 79 |
No data |
c.894G>A/ c.894G>A |
|
Heterozygous for the variant c.894G>A patients |
||||||||
|
1 |
F |
no complain;
hepatomegaly (9m) |
8y |
ALT 198.0 AST 78.7 TC 7.89 HDL 0.70 LDL 6.00 TG n.d. |
Homogeneity and increased echogenicity of liver parenchyma in US;
|
LAL 0.00 |
No data |
c.894G>A/c.894+1G>A |
|
2 |
M |
no complain;
hepatomegaly (3y) |
7y |
ALT 147.5 AST 124.1 TC 9.70 HDL n.d. LDL 7.90 TG n.d. |
Porto-portal fibrosis in US |
LAL 0.02
Cht 274,3 |
No data |
c.894G>A/c.796G>T |
|
3 |
F |
recurrent abdominal pain;
hepatosplenomegaly, elevated transaminases (3y) |
4y |
ALT 150.0 AST 165.4 TC 8.05 HDL n.d. LDL 7.30 TG n.d. |
Decreased echogenicity of liver in US |
LAL 0.01
Cht 485 |
No data |
c.894G>A/c.796G>T |
|
4 |
F |
weakness, tightness of the right hypo-chondrium;
hepatosplenomegaly, elevated transaminases (7y) |
18y |
ALT 122.0 AST 99.0 TC 8.67 HDL 2.07 LDL 5.57 TG 2.26 |
Mildly diffused heterogeneity, increased echogenicity of liver in US
|
LAL 0.00
Cht 1229,6 |
C-triol143
7-KC 650 |
c.894G>A/c.796G>T |
|
5 |
M |
no complain;
hepatomegaly, elevated transaminases (4y) |
14y |
ALT 109.0 AST 82.0 TC 6.18 HDL 0.90 LDL 5.59 TG n.d. |
Diffuse changes of liver parenchyma, features of liver fibrosis in US |
LAL 0.02 Cht 148 |
C-triol75
7-KC 150.6 |
c.894G>A/ c.421delG |
|
6 |
F |
no complain;
elevated transaminases (4y) |
5y |
ALT 128 AST 62 TC 6.18 HDL 1.20 LDL 5.85 TG n.d. |
Diffuse changes of liver parenchyma, features of liver fibrosis in US |
LAL 0.04
Cht 595,5 |
No data |
c.894G>A/ c.796G>T |
|
7 |
M |
no complain;
hepatosplenomegaly, elevated transaminases (7y) |
10y |
ALT 143 AST 207 TC 7.59 HDL 0.80 LDL 4.63 TG 1.76 |
Homogeneity, decreased echogenicity of liver parenchyma in US; |
LAL 0.01
Cht 356 |
C-triol 155
7-KC 405 |
c.894G>A/c.420G>A |
|
8 |
M |
no complain;
mild hepatomegaly, elevated transaminases (3y) |
8y |
ALT 258 AST 183 TC 6.73 HDL 0.59 LDL 5.49 TG 2.20 |
Diffuse changes of liver parenchyma in MRT; |
LAL 0.00
Cht 398 |
C-triol44
7-KC 370 |
c.894G>A/ c.796G>T |
|
9 |
F |
no complain;
diagnosed through family screening, mild hepatomegaly, elevated transaminases (9y) |
9y |
ALT 88 AST 56 TC 9.05 HDL 0.80 LDL 7.77 TG 2.36 |
Diffuse changes of liver parenchyma in US |
LAL 0.02
Cht 67 |
No data |
c.894G>A/c.420G>A |
|
10 |
M |
recurrent abdominal pain;
mild hepatomegaly, elevated transaminases (3y) |
6y |
ALT 132 AST 106 TC 7.04 HDL 0.90 LDL 5.66 TG 1.99 |
Diffuse changes of liver parenchyma in US;
|
LAL 0.01
Cht 45 |
C-triol53.5
7-KC 165 |
c.894G>A/c.420G>A |
|
11 |
F |
recurrent abdominal pain;
hepatosplenomegaly, elevated transaminases, hyper-bilirubinemia (8y) |
12y |
ALT 96 AST 74 TC 9.74 HDL 1.35 LDL 7.94 TG 2.17 |
Small-focal heterogeneity, increased echogenicity of liver parenchyma in US; |
LAL 0.00
Cht 42 |
C-triol59.2
7-KC 167.4 |
c.894G>A/c.817_818delAA |
|
12 |
M |
recurrent abdominal pain;
elevated transaminases, hyperbilirubinemia (3y) |
6y |
ALT 115 AST 111 TC 7.20 HDL 0.80 LDL 5.60 TG n.d. |
Diffuse changes of liver parenchyma, features of fibrosis in MRT; |
LAL 0.00
Cht 165,8 |
C-triol96.8
7-KC 523 |
c.894G>A/c.398C>A |
|
13 |
M |
no complain;
hepatomegaly, elevated transaminases (6m) |
5y |
ALT 406 AST 14 TC 6.63 HDL 0.96 LDL 5.30 TG n.d. |
Diffuse heterogeneity, increased echogenicity of liver in US |
LAL 0.00
Cht 97,9 |
No data |
c.894G>A/c.309C>A |
|
14 |
M |
protracted jaundice, pain in the right hypo-chondrium; hepatosplenmegaly, elevated transaminases, hyperbilirubinemia (18y) |
21y |
ALT 358 AST 47 TC 5.84 HDL 1.56 LDL 3.69 TG 1.29 |
Mild diffuse heterogeneity, increased echogenicity of liver in US
|
LAL 0.00
Cht 43 |
No data |
c.894G>A/ c.309C>A |
|
15 |
F |
no complain;
mild hepatomegaly, elevated transaminases (5y) |
5y |
ALT 178 AST 247 TC 7.27 HDL 1.30 LDL 5.86 TG 1.21 |
Diffuse heterogeneity, increased echogenicity of liver in US; |
LAL 0.01
Cht 89 |
No data |
c.894G>A/ c.911_912delinsT |
|
16 |
M |
enlarged abdomen;
mild hepatomegaly (2y 8m) |
9y |
ALT 40.0 AST 57.0 TC 8.50 HDL 0.57 LDL 6.58 TG n.d. |
Hepatomegaly and increased echogenicity of liver in US
|
LAL 0.00
Cht 884 |
No data |
c.894G>A/ c.796G>T |
|
17 |
M |
recurrent abdominal pain;
hepatosplenomegaly, elevated transaminases (2y) |
7y |
ALT 203 AST 142 TC 9.50 HDL 1.10 LDL 7.09 TG n.d. |
Decreased echogenicity of liver in US |
LAL 0.01
Cht 364 |
C-triol60.6
7-KC 400.1 |
c.894G>A/ c.796G>T |
|
18 |
M |
no complain;
mild hepatomegaly (4y) |
15y |
ALT 78 AST 89 TC 6.25 HDL n.d. LDL 5.10 TG n.d.
|
Homogeneity, decreased echogenicity of liver parenchyma in CT;
Liver biopsy (5y) – periportal fibrosis, lipid storage vacuoles in hepatocyes and Kupffer cells. Liver fibrosis in METAVIR score F2 |
LAL 0.02
Cht 672 |
No data |
c.894G>A/ c.347G>A |
|
19 |
F |
no complain;
mild hepatomegaly, elevated transaminases (3y) |
6y |
ALT 128 AST 79 TC 7.24 HDL 0.71 LDL 5.85 TG 1.20 |
Diffuse heterogeneity, increased echogenicity of liver in US
|
LAL 0.02
Cht 46 |
No data |
c.894G>A/ ?.-3?>G |
|
Patients without c.894G>A variant in the genotype |
||||||||
|
1 |
F |
no complain;
hepatosplenomegaly, elevated transaminases (1y 6m) |
13y |
ALT 308 AST441 TC 11.80 HDL 0.80 LDL 8.98 TG 2.30 |
Liver chirrosisin MRT (14y)
Liver biopsy (16y) – periportal fibrosis liver fibrosis in METAVIR score F-4 (cirrhosis) |
LAL 0.01
Cht 251 |
No data |
c.600G>A/ c.600G>A |
|
2 |
F |
no complain;
mild hepatomegaly (7m) |
19 y |
ALT 129.0 AST 120.0 TC 6.80 HDL 1.46 LDL 4.29 TG n.d. |
Increased echogenicity of liver in US (3y);
Liver biopsy (18y) – periportal fibrosis liver fibrosis in METAVIR score F-4 (cirrhosis) |
LAL 0.00
Cht 99 |
No data |
c.177_180dup4/c.956A>T |
Table 2: Individual characteristics of cholesteryl ester storage disease patients.
ALT - Alanine Aminotransferase Concentration, U/L (<37)
AST - Aspartate Aminotransferase Concentration, U/L (<47)
TC - Serum total cholesterol concentration, mmol/L (3,20-5,20)
HDL - serum High Density Lipoprotein cholesterol concentration, mmol/L (0,90-2,10)
LDL - serum Low Density Lipoprotein cholesterol concentration, mmol/L (1,55-3,80)
TG - serum Triglycerides concentration, mmol/L (0,10-1,70)
LAL - Lisosomal Acid Lipase activity, nmol/punch/h (0,16-1,80)
Cht - Chitotriosidase activity, nmol/punch/h (<100)
C-triol - Cholestane-3β,5α,6β-triol concentration, ng/mL (<50,0)
7-KC - 7-Ketocholesterol Concentration, ng/mL (<75,0)
US - Ultrasound
CT - Computer Tomography
METAVIR F0 - F4 - the stages of fibrosis according to METAVIR scoring system from no fibrosis to cirrhosis respectively, estimated based on histology data
Patients with cholesteryl ester storage disease
The remaining 36 patients demonstrated symptoms of CESD and 34 of them had at least one copy of c.894G>A variant. From the rest two patients, one had a pathogenic variant c.177_180dup4 and an undescribed substitution c.956A>T with doubtful pathogenic features; the second one was homozygous for the variant c.600G>A. The genotypes of patients are presented in table 2.
The age at which the first clinical signs were reported by a physician varied from 4 months to 18 years (median 3 years). Hepatomegaly was the most common first abnormality observed in 28 patients (71.8%), while 9 (23.1 %) patients were noted to have mild splenomegaly. Twenty-seven patients (69.2 %) had also mildly to moderately elevated liver transaminases.
In all patients dyslipidemia type II was presented: serum total cholesterol and LDL-cholesterol were found to be elevated in all cases. In addition, cholesterol levels tended to increase with age. The maximum concentration of total cholesterol amounted 11.8 mmol/L (at the age of 13y), the maximum concentration of LDL - 9.63 mmol/L (17y) (for reference ranges see Table 2). At the background of hypercholesterolemia HDL-cholesterol level was normal in 20 patients and decreased in 12 patients (in 4 cases was not performed). Serum triglycerides concentration was performed for 19 patients and noted to be normal in 11 of them, in 8 patient’s triglycerides level was elevated.
Most of the patients demonstrated increased echogenicity of liver and signs of fibrosis in US, CT and MRT studies. Histological data performed for 6 patients, shows periportal fibrosis and microvesicular steatosis of liver. In 5 of them fibrosis with septa was observed (METAVIR score higher than F-0). Three patients had cirrhosis at the age of 10, 14 and 18 years.
Secondary biomarkers in the CESD patients
It is known, that the presence of the splice-junction mutation c.894G>A alleviates the clinical presentation of LAL-D [5]. On the other hand, the presence of two LOF-mutations in a genotype dramatically worsens the disease course leading to WD phenotype. The majority of variants identified in a compound-heterozygous state with the allele c.894G>A found to be LOF-mutations: nonsense variants, frame-shift deletions and insertions. We hypothesized about a significant impact of LOF-alleles presented in a compound-heterozygous state with the allele c.894G>A on the severity of symptoms in comparison with patients homozygous for the “mild” splice-junction variant.
The clinical data presented in table 2 shows that such features as liver transaminases or US findings varied widely in patients with the same genotype, even in one family. That is why we compared the secondary biomarkers of LAL-D, chitotriosidase activity and oxysterols levels in the groups of homozygotes and compound-heterozygotes for c.894G>A patients, respectively. Increase Cht activity reflects the involvement of liver and spleen macrophages [24]. Increased oxysterols could be considered as secondary to lipid profile alterations.
Cht activity was measured in 15 homozygous and 19 compound-heterozygous patients. The enzyme activity in compound-heterozygous patients was found to be significantly higher than in homozygotes (P<0.05). Therefore, statistic data argues for correlation of the marker of liver injure with a LIPA genotype. The degree of macrophages activation and fibrogenesis depends on the age of the clinical presentation and the time from first symptoms (due to accumulation of the substrate). Regarding that we could assume that all these characteristics of the disease course directly correlate with a patient genotype.
The oxysterols levels were measured in 7 homozygous and 8 compound-heterozygous patients. The significant difference among 7-KC and C-triol levels between the groups was observed (P<0.05). However, it should be remarked that there is o any difference among standard characteristics of lipid profile as total cholesterol and LDL-cholesterol. So, the markers of dyslipidemia do not demonstrate one-valued association with one LOF-mutation of LIPA in patients’ genotypes.
The intervals of the significant different characteristics in the two groups are presented on figure 1.
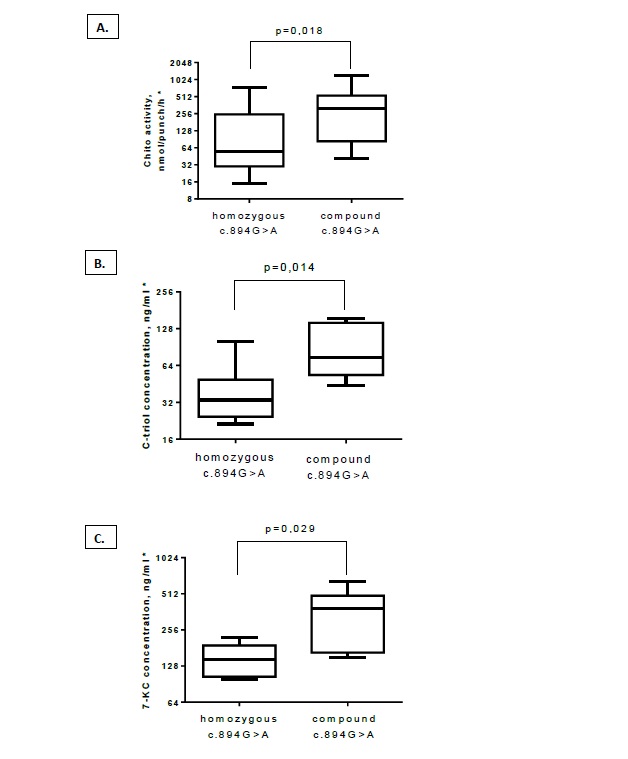 Figure 1: Box plots for secondary biomarkers in the patients with homozygous c.894G>A and compound-heterozygous c.894G>A with LOF-mutations; chitotriosidase activity (A), cholestane-3β, 5α, 6β-triol level (B) and 7-ketocholesterol level (C).
Figure 1: Box plots for secondary biomarkers in the patients with homozygous c.894G>A and compound-heterozygous c.894G>A with LOF-mutations; chitotriosidase activity (A), cholestane-3β, 5α, 6β-triol level (B) and 7-ketocholesterol level (C).
*Logarithmic scale
Regarding a huge progress in the target of LSDs treatment observed in the last years, the clinical significance of their early and correct diagnosis is constantly growing. The problem of LAL-D is non-specific symptoms. The challenging of CESD is the lack of specific symptoms or signs (as we see in WD where there is observed a pathognomonic adrenal gland calcification) and absence of complains in more than half of all reported cases (this ratio is observed in our cohorts too, see table 2) [2].
Measurement of LAL activity in DBS should be considered as one of the mandatory first diagnostic steps in both listed categories. In the case of positive or doubtful results the complex study is needed [5].
The supporting biochemical data are high levels of oxysterols and Cht activity. A quantitative analysis of these biomarkers seems to be useful. The confirmation of the statistic correlations with heterozygous severe LIPA defects we defined is requiring more wide sampling of CESD patients and detailed clinical data. It seems over time measurement to be informative, especially on the background of different treatment courses. If confirmed this data should have diagnostic significance for further detection of c.894G>A and other LIPA variants. It may also have some prognostic meaning, primarily for progression of pathology in the macrophage system of liver related to the Cht activity.
CONCLUSION
The mutation spectrum of LIPA gene in Russian population is varied. We found 16 different pathogenic variants in LAL-D patients with 11 of them undescribed so far.
There were identified two common mutations among the Russian and the Ukrainian patients: c.894G>A variant as the most frequent like in other European countries and c.796G>T variant accounting for 10% of our cohort but comparatively rare in Europe.
Heterozygous LOF-defects of LIPA were found to increase the Cht activity and the levels of the two oxysterols in CESD patients. In comparison with the homozygotes for the “mild” variant c.894G>A, it indicates the higher extent of macrophages activation/liver fibrosis and dyslipidemia progression, respectively. Those data may have a prognostic significance but more detailed studies are required.
ACKNOWLEDGEMENT
The authors would like to thank physicians of all patients providing of medical records and consultation, medical staff of Morozov municipal children’s Hospital of Moscow for plasma sampling, our patients and their families.
CONFLICT OF INTEREST
The authors EA Kamenets, SV Mikhaylova, A Tylki-Szyma?ska, VS Kakaulina, NL Pechatnikova, N Pichkur, P Lipi?ski, GV Baydakova, TY Proshlyakova, IO Nagornov, EY Zakharova declare that they have no conflict of interest.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Reiner Ž, Guardamagna O, Nair D, Soran H, Hovingh K, et al. (2014) Lysosomal acid lipase deficiency--an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis 235: 21-30.
- Bernstein DL, Hülkova H, Bialer MG, Desnick RJ (2013) Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol 58: 1230-1243.
- Ruiz-Andrés C, Sellés E, Arias A, Gort L, Spanish LAL Deficiency Working Group (2017) Lysosomal Acid Lipase Deficiency in 23 Spanish Patients: High Frequency of the Novel c.966+2T>G Mutation in Wolman Disease. J Inher Metab Dis Rep 37: 7-12.
- Klima H, Ullrich K, Aslandis C, Fehringer P, Lackner KJ, et al. (1993) Splice Junction Mutation Causes Deletion of a 72-Base Exon from the mRNA for Lysosomal Acid Lipase in a Patient with Cholesteryl Ester Storage Disease. J Clin Invest 92: 2713-2718.
- Lipi?ski P, ?ugowska A, Zakharova EY, Socha P, Tylki-Szyma?ska A (2018) Diagnostic Algorithm for Cholesteryl Ester Storage Disease: Clinical Presentation in 19 Polish Patients. J Pediatr Gastroenterol Nutr 67: 452-457.
- Valles-Ayoub Y, Esfandiarifard S, No D, Sinai P, Khokher Z, et al. (2011) Wolman disease (LIPA p.G87V) genotype frequency in people of Iranian-Jewish ancestry. Genet Test Mol Biomarkers 15: 395-398.
- Aguisanda F, Thorne N, Zheng W (2017) Targeting Wolman Disease and Cholesteryl Ester Storage Disease: Disease Pathogenesis and Therapeutic Development. Curr Chem Genom Transl Med 11: 1-18.
- Muntoni S, Wiebusch H, Jansen-Rust M, Rust S, Seedorf U, et al. (2007) Prevalence of cholesteryl ester storage disease. Arterioscler Thromb Vasc Biol27: 1866-1868.
- Scott SA, Liu B, Nazarenko I, Martis S, Kozlitina J, et al (2013) Frequency of the cholesteryl ester storage disease common LIPA E8SJM mutation (c.894G>A) in various racial and ethnic groups. Hepatology 58: 958-965.
- Hamilton J, Jones I, Srivastava R, Galloway P (2012) A new method for the measurement of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin Chim Acta 413: 1207-1210.
- vom Dahl S, Harzer K, Rolfs A, Albrecht B, Niederau C, et al. (1999) Hepatosplenomegalic lipidosis: what unless Gaucher? Adult cholesteryl ester storage disease (CESD) with anemia, mesenteric lipodystrophy, increased plasma chitotriosidase activity and a homozygous lysosomal acid lipase -1 exon 8 splice junction mutation. J Hepatol 31: 741-746.
- Pajares S, Arias A, García-Villoria J, Macías-Vidal J, Ros E, et al. (2015) Cholestane-3β,5α,6β-triol: high levels in Niemann-Pick type C, cerebrotendinous xanthomatosis, and lysosomal acid lipase deficiency. J Lipid Res 56: 1926-1935.
- Ageeva NV, Agapova LA, Amelina EL, Gundobina OS, Zharkova MS, et al. (2018) Progressive liver disease: ? deficiency of lysosomal acid lipase (clinical cases). RMJ 5: 96-103.
- Bychkov IO, Kamenets EA, Filatova AY, Skoblov MY, Mikhaylova SV, et al. (2019) The novel synonymous variant in LIPA gene affects splicing and causes lysosomal acid lipase deficiency. Mol Genet Metab 127: 212-215.
- Boenzi S, Deodato F, Taurisano R, Martinelli D, Verrigni D, et al. (2014) A new simple and rapid LC-ESI-MS/MS method for quantification of plasma oxysterols as dimethylaminobutyrate esters. Its successful use for the diagnosis of Niemann-Pick type C disease. Clin Chim Acta 437: 93-100.
- Richards S, Aziz N, Bale S, Bick D, Das D, et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405-424.
- Zakharova EY, Kamenets EA, Baydakova GV, Mikhaylova SV, Strokova TV, et al. (2017) Lysosomal acid lipase deficiency diagnostics and mutation spectrum of lipa gene in cohort of patients with hypercolesterolemia. Atherosclerosis 263: 60.
- Reynders J, Burton BK, del Angel G (2016) Novel LIPA mutations resulting in lysosomal acid lipase deficiency. Mol Genet Metab 120: 17-145.
- Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, et al. (2015) The human splicing code reveals new insights into the genetic determinants of disease. 347: 1254806.
- Vinje T, Wierød L, Leren TP, Strøm TB (2018) Prevalence of cholesteryl ester storage disease among hypercholesterolemic subjects and functional characterization of mutations in the lysosomal acid lipase gene. Mol Genet Metab 123: 169-176.
- Anderson RA, Bryson GM, Parks JS (1999) Lysosomal Acid Lipase Mutations That Determine Phenotype in Wolman and Cholesterol Ester Storage Disease. Mol Genet Metab 68: 333-345.
- Cappuccio G, Donti TR, Hubert L, Sun Q, Elsea SH (2019) Opening a window on lysosomal acid lipase deficiency: Biochemical, molecular, and epidemiological insights. J Inherit Metab Dis 42: 509-518.
- Aslanidis C, Klima H, Lackner KJ, Schmitz G (1994) Genomic organization of the human lysosomal acid lipase gene (LIPA). Genomics 20: 329-331.
- Malaguarnera L (2006) Chitotriosidase: the yin and yang. Cell Mol Life Sci 63: 3018-3029.
Citation: Elena K, Svetlana M, Anna T, Victoria K, Natalia P, et al. (2019) Characteristics of the Phenotype-Genotype Correlation in Cohort of Russian and Ukrainian Patients with Lysosomal Acid Lipase Deficiency. J Genet Genomic Sci 4: 013.
Copyright: © 2019 Kamenets Elena, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.