Complete Androgen Insensitivity Syndrome: Primary Amenorrhoea of Rare Variety
*Corresponding Author(s):
Shirin JahanDepartment Of Reproductive Endocrinology And Infertility, Bangabandhu Sheikh Mujib Medical University, Dhaka, Bangladesh
Email:shirin.dr@gmail.com
Abstract
Androgen insensitivity syndrome is an X linked disorder of male sexual differentiation caused by mutation affecting the androgen receptor gene Xq 11-12 resulting in decreased peripheral responsiveness to circulating androgens, with variable phenotypic expression. The aim of this report was to present a rare case of androgen insensitivity syndrome, its cause, diagnosis and treatment along with review of literature. We described a case of a 16-year-old patient who presented with primary amenorrhea, bilateral labial mass and inguinal hernia. She was confirmed of having androgen insensitivity syndrome after testing for abdominal ultrasound, estimation of relevant hormonal levels, karyotyping and histopathological examination of labial mass and was managed by surgery and counselling.
Keywords
Complete androgen insensitivity syndrome; Karyotyping; Primary amenorrhoea
Introduction
Androgen insensitivity syndrome is an X linked disorder of male sexual differentiation caused by mutation affecting the androgen receptor gene Xq 11-12 resulting in decreased peripheral responsiveness to circulating androgens, with variable phenotypic expression [1]. The clinical phenotypes of AIS are variable and are classified into three main categories: Complete (CAIS), Partial (PAIS) and Mild (MAIS) form. CAIS is indicated when the external genitalia are those of a typical female. Mild androgen insensitivity syndrome (MAIS) is indicated when the external genitalia are those of a typical male, and Partial Androgen Insensitivity Syndrome (PAIS) is indicated when the external genitalia are partially but not fully masculinized. The designations reflect the severity of androgen resistance [2].
CAIS prevalence in 46 XY males is estimated from 1 in 20400 to 1 in 99100 [3]. They may present with a short blind ending vagina, absence of Wolffian duct derived structures like epididymis, vas deferens and seminal vesicles, absence of prostate. At puberty breast development is observed but pubic and axillary hair are absent. Mullerian structures rarely occur in CAIS individuals [4]. In CAIS, the risk of Germ Cell Tumors (GCTs) is considered low and related to age. The estimated risk of gonadal tumors in CAIS gonads was about 0.8%- 22% [5].
Case Report
A 16 years old girl of class ten presented with primary amenorrhea and bilateral labial swelling since birth at OPD of Reproductive Endocrinology and Infertility, BSMMU. Physical examination revealed normal stature for the age, appropriate breast development (Tanner stage III), normal external genitalia, but scanty pubic and axillary hair. Right labial swelling was about 2X2 cm & left one was 3X2 cm in size, firm in consistency, non-tender & mobile up to the deep inguinal ring along with positive cough reflex. On per vaginal examination, vagina was blind and admits about 1.5cm of the tip of the finger. Per rectal examination revealed absence of uterus and cervix (Figures 1-3).
Investigations
|
Name of investigations |
Patient |
Reference |
|
CBC with ESR |
Hb% - 14 gm/dl ESR-10 mm/ 1st hour |
|
|
FBS 2HA 75 gm glucose
|
4.4 mmol/L 6.8mmol/L |
|
|
Serum TSH
|
2.51 micro IU/ml |
|
|
Prolactin |
10.31ng/ml |
|
|
FSH |
6.21mIU/ml |
|
|
LH |
11.65 m IU/ml |
|
|
Testosterone |
324.61 ng/dl |
Normal Male range:200-1200ng Normal female range:20-75ng/dl |
|
Estradiol |
34. 57pg/dl |
Normal female rang:50-300pg/dl Normal male range:20-70pg/dl |
|
USG of lower abdomen |
absence of uterus and ovary |
|
|
Karyotyping |
46XY |
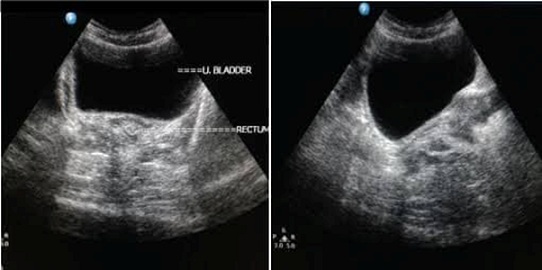 Figure 1: USG showing absence of uterus, cervix and ovary.
Figure 1: USG showing absence of uterus, cervix and ovary.
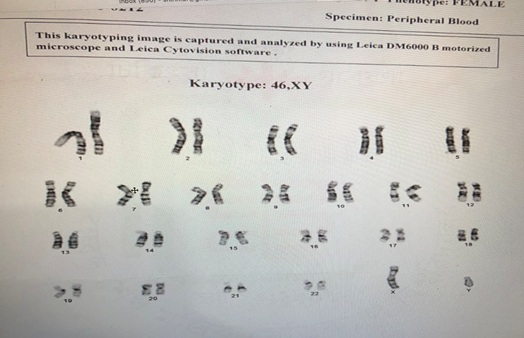 Figure 2: Karyotyping shows 46XY.
Figure 2: Karyotyping shows 46XY.
 Figure 3: Karyotyping report.
Figure 3: Karyotyping report.
Management
After admission, plan of management was made by multidisciplinary approach in collaboration with pediatric surgery department. The patient and parents were counselled about the condition and its prevalence among population, sexual identity, inability of childbearing capacity and the need for surgical removal of the gonads. They were also explained about regular postoperative follow-up, hormone supplementation and vaginal reconstruction. Informed consent was obtained for all the procedures and bilateral gonadectomy. Bilateral crease incisions were made to open the inguinal canal for gonadectomy.
Histopathology revealed two testes with atrophic seminiferous tubules containing only Sertoli cells, associated to a Leydig cells hyperplasia. Hopefully, no signs of testicular cancer were identified. Postoperative evolution was uneventful and the patient left hospital after five days. Estrogen substitution therapy was prescribed to prevent osteoporosis (Figures 4 and 5).
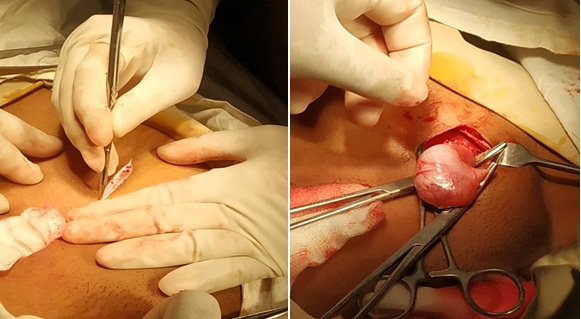 Figure 4: Bilateral crease incision was given to open the inguinal canal for bilateral gonadectomy.
Figure 4: Bilateral crease incision was given to open the inguinal canal for bilateral gonadectomy.
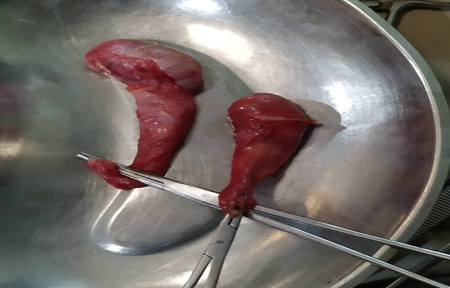 Figure 5: Resected gonads.
Figure 5: Resected gonads.
Discussion
John Morris first described this syndrome, but the exact location of the human AR gene on Xq11-12 locus was discovered in 1989. Test is begin their differentiation at 6th week of male fetal development. These phenomena are under the influence of the SRY gene located on the Y chromosome. Leydig cells start producing testosterone at the 8th week and male sexual characteristics take place including the testicular translocation to the scrotum. Testosterone affects cells through androgen specific nuclear receptors [6]. The affected transactivation domain of the receptor proteins encoded by a gene located on the proximal long arm of the X chromosome, specifically locus Xq11-Xq12 are unable to translate all cells to recognize and use testosterone, thus leading to androgen insensitivity syndrome [7].
All forms of AIS are X-linked traits inherited as a recessive disorder. Individuals affected with CAIS with normal male karyotype (46, XY), have female external genitalia, blind vaginas, an absent uterus and female adnexa, female breast development, and abdominal, inguinal or labial testes. The action of antimüllerian hormone produced by Sertoli cells of the testis results in absence of the uterus, cervix and proximal vagina in complete androgen insensitivity syndrome [8]. Vagina is always blind-ending though varing from a dimple in the perineum to normal length [9]. Diagnosis of CAIS could be done very early by amniocentesis that reported a 46, XY karyotype, and obstetrics ultrasound or clinical evidence at birth showed the presence of female external genitalia.
We found elevated serum levels of testosterone and of LH, due to the androgen insensitivity. Testosterone becomes aromatized, for this reason CAIS patients present with higher estrogen levels than normal male and have good development of breast. Testosterone level of our patient was in normal male level, with high range of serum estradiol. Ultrasound scan confirms the absence of Mullerian structure -uterus, upper part of vagina and absence of both ovaries. Labial swelling ultrasound confirmed the gonads were testis. 46 XY karyotype confirmed our diagnosis. In our case the approach for gonadectomy was inguinal rather than labial because this approach facilitates total hernial tract resection. Following gonadectomy the girl must have long term estrogen replacement therapy to prevent the regression of secondary sexual characteristics and to minimize the consequences of estrogen deficiency.
Conclusion
The Complete form of Androgen Insensitivity Syndrome (CAIS) is very rare. Multidisciplinary approach, psychological and social support are the key points of management of such a girl.
References
- Malipatil V, Malipatil P, Chaitanya VM (2016) Androgen insensitivity syndrome, a case report and literature review. Int J Res Med Sci 4: 1830-1833.
- Galani A, Kitsiou-Tzeli S, Sofokleous C, Kanavakis E, Kalpini-Mavrou A (2008) Androgen insensitivity syndrome: Clinical features and molecular defects. Hormones (Athens) 7: 217-229.
- Oakes MB, Eyvazzadeh AD, Quint E, Smith YR (2008) Complete androgen insensitivity syndrome--a review. J Pediatr Adolesc Gynecol 21: 305-310.
- Quigley CA, De Bellis A, Marschke KB, el-Awady MK, Wilson EM, et al. (1995) Androgen receptor defects: Historical, clinical, and molecular perspectives. Endocr Rev 16: 271-321.
- Pleskacova J, Hersmus R, Oosterhuis JW, Setyawati BA, Faradz SM, et al. (2010) Tumor risk in disorders of sex development. Sex Dev 4: 259-269.
- Morris JM (1953) The syndrome of testicular feminization in male pseudohermaphrodites. Am J Obstet Gynecol 65: 1192-1211.
- Brown CJ, Goss SJ, Lubahn DB, Joseph DR, Wilson EM, et al. (1989) Androgen receptor locus on the human X chromosome: regional localization to Xq11-12 and description of a DNA polymorphism. Am J Hum Genet 44: 264-269.
- Achermann JC, Hughes IA (2012) Disorders of sex development. In: Melmed S, Polonsky KS, Larsen PR, Kronenberg HM (eds.). Williams Textbook of Endocrinology (12thedn). Saunders Elsevier, Philadelphia, USA.
- Hughes IA, Werner R, Bunch T, Hiort O (2012) Androgen insensitivity syndrome. Semin Reprod Med 30: 432-442.
Citation: Jahan S, Akter S, Ishrat S (2022) Complete Androgen Insensitivity Syndrome: Primary Amenorrhoea of Rare Variety. J Reprod Med Gynecol Obstet 6: 091.
Copyright: © 2022 Shirin Jahan, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.