DiGeorge Syndrome in a Newborn - A Diagnostic Challenge
*Corresponding Author(s):
Vasco CarvalhoNeonatal Intensive Care Unit, Department Of Pediatrics, Hospital Of Braga, Braga, Portugal
Tel:+351 967109619,
Email:vascofonsecacarvalho@gmail.com
Abstract
Introduction: DiGeorge syndrome is mainly caused by microdeletion of chromosome 22 (22q11.2) and is characterized by a broad phenotypic spectrum.
Description of case: A 35-year-old healthy primigravida was hospitalized due to preterm labor at 29 weeks and four days. Parents were non-consanguineous with unremarkable family history. Obstetric ultrasound on admission revealed polyhydramnios. A female newborn, was born by caesarean section at 31 weeks and six days due to placental abruption. Respiratory support with Continuous Positive Airway Pressure (CPAP) was started on the fourth day of life due to apneas. Screening echocardiogram showed dilation of aortic root and ascending aorta. Computed tomography coronary angiography confirmed the previous result, also showing a retro-esophageal and retro-tracheal pathway of the right subclavian artery. Several attempts of spontaneous ventilation were made, without success, due to several episodes of apnea (obstructive). A high-pressure oropharyngeal probe and continuous aspiration were placed, allowing spontaneous ventilation on day nine of life. Feeding autonomy gradually improved. DiGeorge molecular study by MLPA (Multiple Ligation dependent Probe Amplification) was requested as facial dysmorphia became more evident in association with the conotruncal cardiac anomaly. A deletion in chromosome 22q11.2 was found. The patient was discharged home at four months and 22 days with multidisciplinary follow-up.
Conclusion: With this clinical case we intend to highlight the importance of a high index of suspicion in the early identification of DiGeorge Syndrome even in the absence of the main clinical manifestations in order to allow an early multidisciplinary orientation, as well as to provide genetic counseling to the patient's relatives.
Keywords
22q11 Deletion syndrome; Combined syndromic immunodeficiencies; DiGeorge syndrome
INTRODUCTION
DiGeorge Syndrome (DGS) is mainly caused by the microdeletion of chromosome 22 (22q11.2), being characterized by a broad phenotypic spectrum [1-10].
The microdeletion of chromosome 22q11.2 results in maldevelopment of both the third and fourth pharyngeal pouches and it is the most common interstitial deletion syndrome, affecting approximately 1 in every 2.000-6.000 live births, with males and females being equally affected [3-5, 8-13].
Approximately 90 percent of the patients with DGS have heterozygous deletions in chromosome 22q11.2 and typically result from de novo microdeletions, therefore supporting a high rate of spontaneous deletion. It is important to add that about 10% of the cases are familial, with autosomal dominant inheritance [4,10,11].
Despite being characterized by a set of malformations, the main features of DGS include conotruncal cardiac anomalies, hypoplastic thymus and parathyroid hypoplasia or aplasia. Nevertheless, a broader spectrum is what characterizes both the presence and the severity of individual features, the latter being individually and independently assessed [3-7,9,11]. Overall, it is extremely important to assess the presence and the extent of abnormalities upon diagnosis, since each one is going to be managed in collaboration with the appropriate medical specialty [3-7,10].
Infants with DGS may have some or all the features described in table 1.
|
Phenotypic features |
Frequency (%) |
|
Cardiac anomalies Interrupted aortic arch Truncus arteriosus Tetralogy of Fallot Atrial or ventricular septal defects Vascular rings |
49 to 83 |
|
Immune deficiency T-cell lymphopenia Delayed IgG production Thymic aplasia with absent T cells |
1 to 77 |
|
Palatal anomalies Velopharyngeal insufficiency Submucous cleft palate Cleft palate Bifid uvula |
69 to 100 |
|
Feeding and swallowing issues |
35 |
|
Speech delay |
79 to 84 |
|
Learning disabilities |
45 to 90 |
|
Developmental delay |
75 |
|
Ophthalmologic abnormalities Posterior embryotoxic (anterior segment dysgenesis) Tortuous retinal vessels Strabismus Ptosis |
7 to 70 |
|
Endocrine Hypocalcemia Growth hormone deficiency |
60 |
|
Psychiatric problems Generalized anxiety Phobias Attention deficit hyperactivity disorder Autism Schizophrenia |
9 to 50 |
|
Skeletal abnormalities |
17 to 19 |
|
Renal abnormalities Structural renal anomaly Renal agenesis Dysplastic kidneys Hydronephrosis |
36 to 37 |
|
Hearing loss Conductive hearing loss Sensorineural hearing loss |
2 to 31 |
|
Neurologic Microcephaly Idiopathic seizures Hypotonia |
8 |
|
Dental issues Caries Enamel hypoplasia Delayed dental eruption |
2,5 |
|
Facial dysmorphism Hypertelorism Antimongoloid slant of the eyes Ear malformation Peculiar mouth Hooded eyelids Bulbous nose tip Broad nasal bridge Posteriorly rotated ears Simple ear helices Micrognathia |
12 to 60 |
Table 1: Clinical findings in patients with chromosome 22q11.2 deletion [2,10,12].
Cardiac anomalies are often discovered in prenatal or neonatal period [10]. Aplasia or hypoplasia of the thymus leads to immunological defects with different levels in terms of severity. Most patients with DGS have mild defects in terms of T cell number, not being regarded as clinical immunodeficient [10,13]. Hypoparathyroidism is observed in half of the patients and its symptoms can be quite severe, with tetany and seizures, or subtle which are related to feeding difficulties, stridor and fatigue.
The clinical manifestations that are suggestive of DGS vary with age. Overall, some of the initial signs of DGS may only include polyhydramnios [2,4,5,10]. In the first days or weeks of life, feeding and swallowing problems with consequent nasal regurgitation may be critical. During infancy or childhood, some of the most typical symptoms include a combination of congenital heart defects, chronic infection, hypernasal speech, hypocalcemia, developmental and language delay, behavioral differences and learning disabilities. Less frequently, some of the symptoms may include genitourinary and gastrointestinal problems, respiratory tract disorders, such as subglottic stenosis, laryngomalacia, tracheomalacia and/or bronchomalacia, hypothyroidism, short stature, skeletal abnormalities, thrombocytopenia, conductive and sensorineural hearing loss - occasionally with cochlear abnormalities, microcephaly, idiopathic seizures and hypotonia. During adolescence and adulthood, cognitive deficits and neuropsychiatric illness (such anxiety disorders and schizophrenia) can also be present [4,5,10,11].
The occult submucosal cleft palate and velopharyngeal dysfunction make the diagnosis more challenging, especially in the early neonatal period.11 Palate anomalies may lead to an increase in feeding difficulties, while velopharyngeal insufficiency may result from structural causes, such as the cleft palate, and/or from neuromuscular problems, such as hypotonia and upper airway obstruction [1]. These anomalies can also lead to swallowing dysfunction and respiratory tract disorders, which imply important clinical consequences [1-6]. Hence, paying attention to these problems is essential, considering that they should be detected and treated as early as possible, especially to avoid more severe complications.
In fact, 22q11.2 deletion is the second most common cause of congenital heart disease and developmental delays, and the most common cause of syndromic palatal anomalies at birth [4,5,12].
It is noteworthy to mention that the diagnosis of DGS can be quite difficult, namely due to the number of potential symptoms and to the variation in phenotypes between individuals. Nevertheless, DGS seems to be frequently suspected in patients with one or more signs of deletion. In this case, a diagnosis of 22q11.2 is confirmed, namely by using the Fluorescence in Situ Hybridization (FISH). Still, some newer methods of analysis include the Multiplex Ligation-dependent Probe Amplification Assay (MLPA) and the quantitative Polymerase Chain Reaction (qPCR), which are both able to detect the atypical deletions in 22q11.2 that are not detected by the FISH method [2,5,11].
Both the management and the long-term follow-up of a newborn with DGS require an individualized, multidisciplinary and coordinated care plan, which must also consider the specific clinical manifestations of the patient [5,7,10]. We present a clinical case of a newborn with DGS, with a challenging diagnosis, in which classical manifestations of DiGeorge Syndrome were initially lacking.
CLINICAL CASE
First gestation of a healthy, non-consanguineous parents, with unremarkable family history. Both the first and the second trimester prenatal ultrasound were normal. The mother, 35 years old, was hospitalized at 29 weeks and four days, at risk of preterm labor with polyhydramnios being detected. Tocolytic drugs, magnesium sulphate and fetal lung maturation were administered. The mother gave birth at 31 weeks and six days to a female newborn by caesarean section due to placental detachment. At birth, the infant had Apgar scores of six and ten, in the first and fifth minute, respectively, requiring positive pressure ventilation. Continuous Positive Airway Pressure (CPAP) was started with a Positive End-Expiratory Pressure (PEEP) of 5 Centimeters of water (cmH2O), and with a supplemental Fraction of Inspired Oxygen (FiO2) of 0.3, with a fast recovery over the first hours after birth. The anthropometric measurements, according with Fenton preterm growth chart, were weight 1575 grams (P10-50), length 39cm (P10-50) and head circumference 30cm (P50-90). Physical examination was normal. On the fourth day, CPAP was restarted (PEEP: 5cmH2O, FiO2 21%) due to the accumulation of abundant oropharyngeal secretions with obstructive apneas (Figure 1) and hypoxemia. A chest radiograph was performed and had no apparent alterations.
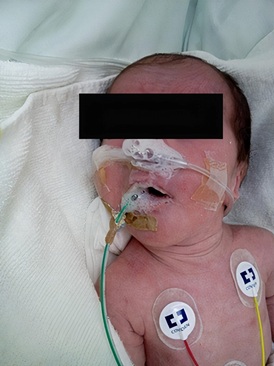 Figure 1: Accumulation of abundant oropharyngeal secretions that led to severe apneas.
Figure 1: Accumulation of abundant oropharyngeal secretions that led to severe apneas.
Several unsuccessful attempts to spontaneous ventilation were made, due to obstructive apneas followed by multiple episodes of severe desaturations due to the presence of abundant oropharyngeal secretions.
Screening Echocardiography showed a dilation of the aortic root and the ascending aorta. A Computerized Tomography coronary angiography (CT coronary angiography) confirmed the previous result, and also showed a retro-esophageal and retrotracheal pathway of the right subclavian artery (Figure 2). Despite this anatomical alteration, cardiology evaluation showed normal cardiac function.
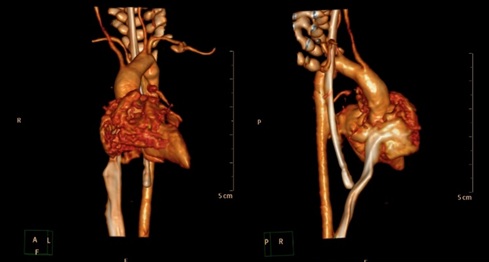 Figure 2: A CT coronary angiography showing the retro-esophageal and retrotracheal pathway of the right subclavian artery.
Figure 2: A CT coronary angiography showing the retro-esophageal and retrotracheal pathway of the right subclavian artery.
This result raised the suspicion of an extrinsic compression, by the anomalous vessel, which could justify the obstructive apneas. An oropharyngeal probe was placed in a high position, in continuous aspiration, which allowed for spontaneous ventilation after day nine.
During hospitalization, facial dysmorphismwhich associated with conotruncal anomaly, raised the suspicion for DGS. A MPLA study for DiGeorge was requested and revealed 22q11.2 microdeletion (MIM#188400), therefore corroborating the suspicion. Both parents were also tested, not presenting any microdeletion. The chest radiograph was reassessed showing absence of the thymic shadow, which had not been perceived in the first imaging evaluation (Figure 3).
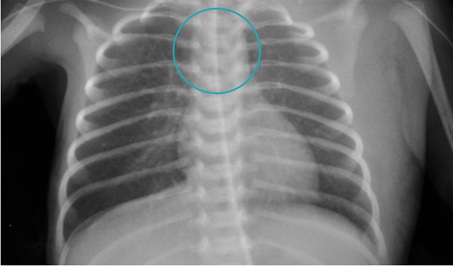 Figure 3: Absence of the thymic shadow in the thoracic chest radiograph.
Figure 3: Absence of the thymic shadow in the thoracic chest radiograph.
During hospitalization, the newborn had no infectious intercurrences, presenting a normal immunological study. She had an immunodeficiency pediatric consult, being kept under clinical and analytical follow-up. She remains without any infections and presenting normal values in immunological evaluations.
During the neonatal period there was no record of tetany or seizures. The laboratory evaluation showed no hypocalcemia or elevated serum phosphorus, and the thyroid and parathyroid hormone levels were normal. A renal-pelvic ultrasonography was performed in order to exclude genitourinary alterations, but no alterations were found.
Given the fact that she needed continuous aspiration of secretions, velopharyngeal incompetence was suspected. Several exams were conducted, namely by otorhinolaryngology, pneumology and gastroenterology. A flexible nasofibrolaryngoscopy was requested, which confirmed the velopharyngeal insufficiency, and revealed hypogenesis of the uvula and a submucosal cleft of the posterior third of the palate. A maxillofacial computed tomography confirmed the submucosal cleft, as well as the asymmetry of the soft palate and the elevation of the hard palate. A Fiberoptic Endoscopic Examination of Swallowing (FEES) showed abundant reflux of milk to the nasopharynge (velopharyngeal incompetence) and a poor propulsion. A bronchoscopy was performed detecting salivary stasis at the level of the larynx.
To improve the velopharyngeal competence, speech and occupational therapy were started. No improvement was perceived so a Percutaneous Endoscopic Gastrostomy (PEG) was placed. The consequent improvement in the accumulation of secretions led to the cessation of the need for continuous aspiration.
Upon discharge, she was four months and 22 days (chronological age of two months and three weeks), and a multidisciplinary follow-up was guaranteed. Currently, she is 4 years and six months old, being clinically stable and not presenting sensorineural hearing loss. Nevertheless, she presents a delay in communication.
She still needs frequent aspiration of the secretions and due to the increase in upper airway resistance at night, requires a CPAP (PEEP of 6 cm H2O with a FiO2 0.21), with no record of apneas or desaturations. She attended the pediatric emergency department several times due to respiratory infections, some of them resulting in hospitalizations due to hypoxemia.
A new FEES confirmed an improvement of swallowing function. However, she still needs PEG, especially for liquids.
DISCUSSION WITH CONCLUSION
With this case we aim to emphasize the importance of having a high index of suspicion in order to early identify DGS even when some of the main clinical manifestations of the DGS are lacking. As there are no pathognomonic manifestations of the syndrome, it is important to recognize that the main features may vary depending on the patient’s age [5,6].
The classic triad of DGS is related to conotruncal cardiac anomalies, hypoplastic thymus, and hypocalcemia. However, and according to the existing literature, the phenotype is quite variable, as it is pointed out in the present clinical case [1-5,12].
In this case report, her main clinical manifestations were due to velopharyngeal incompetence: Polyhydramnios, obstructive apneas and feeding difficulties. Initially such symptoms were considered to be the result of the vascular ring (retro-esophageal and retrotracheal pathway of the right subclavian artery). However, the degree of the tracheal and esophageal compression was not critical enough to cause airway obstruction or feeding and swallowing issues. As the newborn grew, new symptoms became evident, such as facial dysmorphia which, associated with cardiac anomalies and with the maintained velopharyngeal incompetence, increased the suspicion of this syndrome.
The palatal defects were not so obvious on standard physical exam and otolaryngologic findings were decisive to better characterize the degree of velopharyngeal incompetence. At an early stage the predominant clinical symptoms of the newborn were the abundant secretions and the consequent obstructive apneas. After PEG placement there was clinical improvement. At follow-up, despite PEG, she still presents some feeding difficulties. It has been reported that PEG placement should be considered for patients that will require some supplementation for longer periods of time, but should be removed when no longer needed [2,5,6].
According to literature, polyhydramnios is also a clinical feature associated with 22q11.2 deletion syndrome, as seen in our clinal case but it was only reported on obstetric ultrasound at admission [4,10]. So it may serve as an important prenatal clue to the diagnosis, providing a postnatal alert, especially when accompanied by cardiovascular abnormalities. These early manifestations may lead to a suspicion and therefore, to the diagnosis, by performing the genetic test [4,5].
A spectrum of thymic abnormalities is possible in DGS, even though its nature in such patients is not entirely clear [5]. It has been reported that the absence of thymic shadow in chest X-ray is not accurate in predicting individual immune function, as it does not correlate to the size, absence or presence of thymic tissue. Small rests of thymus can be hidden in the neck or mediastinal structures. Thus, the immune system requires specific laboratory evaluation [5,13]. Our clinical case illustrates this aspect. After the diagnosis of DGS, the chest X-ray was reevaluated, demonstrating that the thymic shadow was absent, which motivated an immunological study that presented normal results. During hospitalization the child had no infectious intercurrences. During newborn period she never had hypocalcemia, although hypocalcemia is present in up to 60 percent of the newborns [1-5].
After being discharged, she was evaluated several times in the pediatric emergency department, mainly due to recurrent pulmonary infections with hypoxemia. This clinical case represented a challenging diagnosis. An early recognition of the DGS patients is extremely important in order to provide an early multidisciplinary evaluation, contributing to decrease the healthcare costs [1-6,11,12].
Nevertheless, this clinical case also aims to highlight the need of a genetic counseling to family members, in order to decrease their emotional distress and to inform about the possibility of recurrence (low in a case with a de novo deletion) and the availability of prenatal diagnosis in future pregnancies [11].
REFERENCES
- Jones JW, Tracy M, Perryman M, Arganbright JM (2018) Airway anomalies in patients with 22q11.2 deletion syndrome: A 5-year review. Ann Otol Rhinol Laryngol 127: 384-389.
- McDonald-McGinn DM, Sullivan KE (2011) Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine 90: 1-18.
- Rayannavar A, Katz LEL, Crowley TB, Lessig M, Grand K, et al. (2018) Association of hypocalcemia with congenital heart disease in 22q11.2 deletion syndrome. American Journal of Medical Genetics Part A 176: 2099-2103.
- Sacca R, Zur KB, Crowley TB, Zackai EH, Valverde KD, et al. (2017) Association of airway abnormalities with 22q11.2 deletion syndrome. Int J Pediatr Otorhinolaryngol 96:11-14.
- McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, et al. (2015) 22q11.2 deletion syndrome. Nat Rev Dis Primers 1: 1-19.
- Verheij E, Speleman L, Molen ABM, Thomeer HGXM (2018) Congenital respiratory tract disorders in 22q11.2 deletion syndrome. International Journal of Pediatric Otorhinolaryngology 104: 1-4.
- Cancrini C, Puliafito P, Digilio MC, Soresina A, Martino S, et al. (2014) Clinical features and follow-up in patients with 22q11.2 deletion syndrome. J Pediatr 164: 1475-1480.
- Lindsay EA (2001) Chromosomal microdeletions: Dissecting del 22q11 syndrome. Nature Reviews Genetics 2: 858-868.
- Kraus C, Vanicek T, Weidenauer A, Khanaqa T, Stamenkovic M, et al. (2018) DiGeorge syndrome: Relevance of psychiatric symptoms in undiagnosed adult patients. The Central European Journal of Medicine 130: 283-287.
- Sullivan K (2019) Chromosome 22q11.2 deletion syndrome and DiGeorge syndrome. Immunological Reviews 287: 186-201.
- Barry JC, Crowley TB, JyonouchiS, Heimall J, ZackaiEH, et al. (2017) Identification of 22q11.2 deletion syndrome via newborn screening for severe combined immunodeficiency. Journal of Clinical Immunology 37: 476-485.
- Tang KL, Antshel KM, Fremont WP, Kates WR (2015) Behavioral and psychiatric phenotypes in 22q11.2 deletion syndrome. Journal of Developmental & Behavioral Pediatrics 36: 639-650.
- Morsheimer M, Whitehorn TFB, Heimall J, Sullivan KE (2017) The immune deficiency of chromosome 22q11.2 deletion syndrome. American Journal of Medical Genetics Part A 173: 2366-2372.
Citation: Carvalho V, Oliveira R, Vilaça J, Rocha MG, Pereira A, et al. (2020) DiGeorge Syndrome in a Newborn - A Diagnostic Challenge. J Neonatol Clin Pediatr 7: 061.
Copyright: © 2020 Vasco Carvalho, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.