Disrupting Spinal Caveolin-1 Scaffolding Prevents as well as Reverses Tolerance to Opioid Antiallodynia in Treating Neuropathic Pain in Rats
*Corresponding Author(s):
Nai-Jiang LiuDepartment Of Obstetrics And Gynecology, SUNY Downstate Medical Center, 450 Clarkson Ave, Brooklyn, NY 11203, United States
Email:nai-jiang.liu@downstate.edu
Abstract
Pharmacodynamic opioid analgesic tolerance, resulting in escalating doses of opioids, and paradoxical opioid-induced nociception, remains a major impediment to proscribing opioids for chronic pain management, often leading to undermanaging pain and deleterious consequences thereof. To date, none of the many downstream cellular adaptations proposed to be causally associated with opioid tolerance have reached clinical relevance and identification of underlying upstream mechanism(s) common to most, if not all tolerance adaptations, has remained elusive. We predicted that altered functionality of the predominant scaffolding protein Caveolin 1 (Cav1) is an upstream mechanism fundamental to the developmental and maintenance of morphine anti-allodynic tolerance based on our earlier finding that opioid receptors are present in a macromolecular signaling complex whose formation likely requires a scaffolding protein(s) as well as the ability of chronic opioids to orchestrate functionally interrelated adaptations among signaling molecules. Findings obtained in rats utilizing the spinal nerve ligation neuropathic pain model demonstrated that (1) persistent interference with spinal Cav1 scaffolding and/or spinal Src kinase activity (which is activated by opioids and modulates Cav1 functionality via its phosphorylation) abolished the development of spinal morphine anti-allodynic tolerance and (2) acute disruption of spinal Cav1 scaffolding and/or spinal Src activity after tolerance was fully manifest reinstated opioid naïve anti-allodynic effects of intrathecal morphine. These findings indicate the relevance to pain management of developing anti-opioid tolerance pharmacotherapies that disrupt Cav1 scaffolding in the presence of chronic opioids. The clinical success of such opioid adjuvants is foreshadowed by the potential to concomitantly interrupt multiple analgesic/anti-allodynic tolerance mechanisms.
Keywords
Anti-allodynia; Caveolin 1; Chronic pain; Opioid tolerance; Spinal nerve ligation
Introduction
Opioids continue to be amongst the most efficacious drugs for pain management, being essentially indispensable for mitigating pain that is unresponsive to non-pharmacological treatments and non-opioid analgesics [1]. However, despite their clinical usefulness, pharmacodynamic opioid analgesic tolerance, i.e., classical pharmacological opioid analgesic tolerance, which is independent of disease progression [2-7], is a major contributor to the need for escalating opioid doses during chronic pain management and thus to the current epidemic of death by opioid overdose. This substantially compromises the clinical utilization of opioids, leading to undermanaging nociception, which itself can have substantial damaging consequences [8-13], a poignant examplar of which is that minimizing nociception following surgery is a critical determinant of speed of recovery [14].
µ-Opioid receptor (MOR) downregulation and G protein uncoupling [15-18] have been thought to be foundational to opioid analgesic tolerance. However, many observations challenge this perspective, e.g., (a) substantial opioid analgesic tolerance develops without accompanying MOR downregulation [19-22], MOR G protein uncoupling [23], arrestin binding and/or endocytosis [24,25]; (b) development of opioid analgesic tolerance is often accompanied by upregulation of MOR density [26-31], increased efficacy of MOR agonists [32], and enhanced MOR Gs coupling/Adenylyl Cyclase (AC)/Protein Kinase A (PKA) signaling [31,33-46].
Strikingly, chronic morphine induces coordinated, interrelated changes in signaling molecules downstream from MOR. These include: (a) adaptations that collectively shift MOR signaling from AC inhibitory to stimulatory [33-35,38,40,42,]; (b) increased expression/translocation of a panoply of signaling molecules, e.g., Protein Kinase Cg (PKCg) [47-49], Mitogen Activated Protein Kinase (MAPK) [50-52], Ca2+/calmodulin-dependent protein Kinase IIa (CamKIIa) [53], mGluR5 [54], as well as reciprocal modulation of two phospholipase C isoforms (b1 and b3) [37]. The manifestation of synchronized, functionally interconnected alterations in signaling molecules in response to chronic morphine, along with our recent finding that MOR and the kappa-opioid receptor exist, at least in part, within a common macromolecular signaling complex [55,56] suggests that scaffolding proteins likely play a pivotal role in acute as well as chronic opioid sequelae.
One such scaffolding protein is Caveolin 1 (Cav1), the principal structural protein of caveolae, membrane microdomains [57,58] containing a wide spectrum of signaling proteins [e.g., AC, MAPK, Src, functional G protein-coupled receptors [57,59,60]. This enables Cav1 to spatially and temporally organize signaling complexes, thereby modulating their interactions [61-64]. Importantly, Cav1 can form scaffolds outside of as well as within caveolae [65,66], which also regulate interactions among plasma membrane signaling proteins. This is notable since CNS neurons do not contain structurally defined caveolae [67], but do contain Cav1 [66-72].
Cav1 influences on signaling vary, depending on linked signaling partners, cellular context and the nature of specific signaling pathways. For example, Cav1 suppresses Extracellular signal-Regulated Kinase (ERK)-induced activation of the Grb2-Sos-Ras pathway [73], but facilitates ERK activation of the PI3 kinase pathway and insulin receptor signaling [74,75]. Cav1 scaffolding functionality is also dependent on its phosphorylation. The nonreceptor tyrosine kinase Src, whose activity is augmented by MOR agonists [76], phosphorylates Cav1 at tyrosine 14 (pY14Cav1) [77-79], increasing Cav1 recruitment of signaling molecules [79-83]. This pliability of Cav1 scaffolding and its modulation by Src phosphorylation makes both ideally suited to play a pivotal role in differentially altering interactions among signaling molecules, facilitating their assembly into signaling complexes, altering the relative predominance of signaling pathways, as we and others have demonstrated occurs in response to chronic opioids [35,39,44,46,84-86]. Chronic morphine augments Cav1 scaffolding [87] and expression in spinal cord [86] underscores its likely relevance to opioid anti-allodynic tolerance mechanisms.
Accordingly, this study tests the hypothesis that the scaffolding functionality of Cav1 is critical to both the development and maintenance of opioid anti-allodynic tolerance. Findings obtained utilizing the spinal nerve ligation chronic pain model (SpNL) demonstrated that chronic interference with spinal Cav1 scaffolding and/or chronic pharmacological inhibition of spinal Src activity prevented the onset of spinal morphine anti-allodynic tolerance. Strikingly, acute blockade of spinal Cav1 scaffolding and/or acute pharmacological inhibition of spinal Src activity fully reversed tolerance to the anti-allodynic effects of morphine after tolerance had been fully developed. These findings imply the clinical utility of developing opioid adjuvants that target spinal Cav1 and/or spinal Src to minimize tolerance to opioids in pain mitigation. This would attenuate the need for opioid dose escalation, facilitating the use of opioids in pain management.
Methods
Animals and Housing
Experiments utilized male Sprague-Dawley rats (Charles River Laboratories, Kingston, NY; 250-300 g) that were maintained in a controlled environment on a 12-hour light/dark cycle. Food and water were available ad libitum. Our Institutional Animal Care and Use Committee reviewed and approved all experimental procedures, which conformed to applicable national/international guidelines. Animals were randomly assigned to experimental and control groups. Each animal was used only once. Power analysis guided experimental group size, which is indicated in relevant sections of Results.
Spinal Nerve Ligation (SpNL)
As we and others have reported [88-94], an incision was made above the lumbar spine. The left transverse process of vertebra L6 was exposed under isoflurane anesthesia (2.5%). The left L5 spinal nerve was tightly ligated with silk thread No. 6 and cut distal to the ligation. Peripheral neuropathic pain arises within 24 h in the ipsilateral hind paws, persisting for months [88,89,93,94], which is displayed as mechanical allodynia. General behavior of the rats was monitored before and after the surgery. Rats showing difficulty elevating the hind paw ipsilateral to SpNL were excluded from the study.
Implantation of intrathecal cannulas
A permanent indwelling cannula was inserted into the lumbar spinal cord subarachnoid space as we have described and utilized previously for spinal drug delivery [55,56,94]. Spinal cannula was implanted concomitant with SpNL. Briefly, animals were anesthetized as described above, and an 8.0 cm PE-10 catheter (Becton Dickinson and Company, Franklin Lanes, and NJ) was inserted into the spinal subarachnoid space via the atlanto-occipital membrane. The cephalic portion of the catheter was externalized through the skin on the dorsal side of the neck, where it was secured in place, being relatively inaccessible to the paws., All animals utilized in studies appeared to be free of infection upon gross inspection and did not exhibit any motoric impairment, assessed using the righting reflex and the inclined plane test.
Intrathecal administration of drugs
Drugs were applied to the spinal cord subarachnoid space over a 60-second period via the indwelling i.t. cannula seven days following cannula implantation. Flushing the cannula with an additional 10 µl of saline ensured complete delivery. I.t. pretreatment with either caveolin domain competing peptide (CSD), scrambled caveolin domain competing peptide (S-CSD), PP2, PP3 or CSD+PP2 was applied 45 min before i.t. application of morphine. I.t. morphine dose responsiveness was determined 30 min after each i.t. application of morphine, immediately before the application of the following dose of morphine [95-97]. Dose responsiveness was always performed in the identical ascending pattern among the various experimental groups, minimizing any potential confounds resulting from differences in duration of action among the various ascending doses of i.t. morphine.
Chronic morphine administration generating spinal or systemic morphine tolerance
Spinal morphine tolerance was achieved via i.t. application of 15 μg morphine daily for at least six days [98,99]. Systemic morphine tolerance was accomplished by implanting morphine pellet subcutaneously under isoflurane anesthesia (2.5%), one pellet on day 1, two pellets on day 3 and three pellets on day 5 (each containing 75 mg morphine base) [87,100].
Quantification of allodynia and acute thermal nociception
Mechanical allodynia was quantified by measuring withdrawal thresholds of the hind paw ipsilateral to the SpNL in response to the application of von Frey force as we described previously [101]. Briefly, rats were placed on a wire mesh surface, covered by an inverted plastic cage and allowed to habituate for 15 min. A hand-held probe containing a rigid filament was applied to the plantar surface of the hind paw with increasing force. The applied pressure (g) that elicited paw withdrawal was automatically recorded using Electronic von Frey Anesthesiometer (IITC Life Science, Woodland Hills, CA). A cutoff force of 60 g was employed to prevent tissue damage. Tail flick latency in response to the application of radiant heat applied to the tail using an Algesia Meter (IITC, Woodland Hills, CA) was used to assess acute nociception and its modulation by i.t. morphine as we described previously [102]. Intensity of the radiant heat was adjusted such that baseline values were in the range of 3.0-4.5 sec. Tissue damage was prevented by imposing a 10 sec cutoff. All testing was performed blind to treatment.
Drugs
CSD (H-Arg-Gln-Ile-Lys-Ile-Trp-Phe-Gln-Asn-Arg-Arg-Met-Lys-Trp-Lys-Lys-Asp-Gly-Ile-Trp-Lys-Ala-Ser-Phe-Thr-Thr-Phe-Thr-Val-Thr-Lys-Tyr-Trp-Phe-Tyr-Arg-OH) and S-CSD (H-Arg-Gln-Ile-Lys-Ile-Trp-Phe-Gln-Asn-Arg-Arg-Met-Lys-Trp-Lys-Lys-Trp-Gly-Ile-Asp-Lys-Ala-Ser-Phe-Thr-Thr-Phe-Thr-Val-Thr-Lys-Tyr-Trp-Phe-Arg-Tyr-OH) were obtained from Millipore Corporation (Bedford, MA). 3-(4-chlorophenyl) 1 (1,1-dimethylethyl)-1H-pyrazolo [3,4-d]pyrimidin-4-amine (PP2, a Src kinase inhibitor) and 1-Phenyl-1H-pyrazolo [3,4-d]pyrimidin-4-amine (PP3, a negative control for PP2) was obtained from Tocris (Ellisville, MO). Morphine sulfate and 75 mg morphine base pellets were obtained from NIDA. CSD (up to 10 µg), S-CSD (10 µg), PP2 (300 ng) and PP3 (300 ng) were solubilized in Dimethyl Sulfoxide (DMSO, up to 5 µl) while morphine sulfate (up to 20 µg) was prepared in 5 µl saline.
Data analysis
Only-way ANOVA was used to compare treatment effect within group and two-way repeated measure ANOVA was used to compare treatment by time/dose effects among groups. Bonferroni post hoc test identified specific groups between which significant effects were observed. P < 0.05 was considered significant. Statistical comparisons were made using Prism 6 software. Data are expressed as the mean +/- Standard Error of the Mean (SEM).
Results
Combined Intrathecal (i.t.) treatment with CSD and PP2 prevents development of spinal opioid anti-allodynic tolerance induced by chronic i.t. morphine. In these experiments, spinal opioid analgesic tolerance was achieved by the i.t. application of 15 µg morphine, once daily for six days. Figure 1A illustrates that this spinal treatment produced substantial tolerance, essentially eliminating anti-allodynic responsiveness to i.t. morphine (15 µg), assessed using the electronic von Frey test. By day 3, roughly half the initial anti-allodynic effect of i.t. morphine was no longer manifest and by day 6, essentially the totality of i.t. morphine responsiveness had been eliminated. In striking contrast, there was no loss of morphine’s effectiveness in chronic morphine-treated animals that had been pretreated intrathecally with CSD+PP2 (10 µg and 300 ng, respectively), 45 min prior to i.t. morphine. Two-way ANOVA revealed a significant treatment effect (i.t. CSD+PP2 vs. vehicle; n=10 and 6, respectively) (F1,14=92.3, p < 0.0001) after day 2 of i.t. morphine (p < 0.0001 for days 3-6).
Figure 1B illustrates that the totality of the rightward shift of i.t. morphine dose-responsiveness (2.5-20 µg) was eliminated by pretreatment with CSD+PP2, revealing that their tolerance abating effect was not limited by the dose of i.t. morphine. Two-way ANOVA showed a significant treatment effect among the 3 groups: F2,15=54.11, p < 0.0001). Significant differences were observed between Chronic Morphine (CM) group (n=5) and CSD+PP2+CM group (n=6) as well as between CM group and morphine naïve group (n=7) (p < 0.0001 for both comparisons), but not between CSD+PP2+CM and opioid naïve animals (p>0.05). The latter comparison indicates that pretreatment with i.t. CSD+PP2 abolished development spinal morphine anti-allodynic tolerance. This effect of i.t. CSD+PP2 results specifically from its anti-tolerance action since (a) i.t. treatment with vehicle (DMSO) had no effect on tolerance development and (b) as shown in Figure 1C, chronic i.t. treatment with CSD+PP2 did not alter the anti-allodynic effect of acute i.t. morphine in the absence of chronic spinal morphine treatment (F1,18=3.4, p>0.05, n=4 for each point), i.e., i.t. CSD+PP2 neither directly influenced basal von Frey thresholds nor spinal morphine anti-allodynic responsiveness. Instead i.t. CSD+PP2 specifically interrupted mechanisms underlying opioid tolerance development, enabling i.t. morphine to produce normative anti-allodynia despite the chronic morphine treatment (Figure 1).
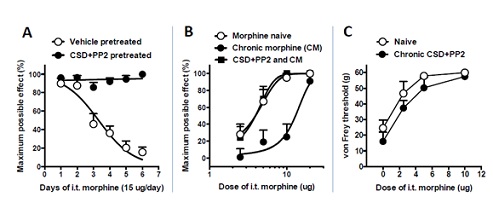 Figure 1: I.t. CSD and PP2 block tolerance development to i.t. morphine. Panel A: Opened circle (control group) illustrates the complete loss of anti-allodynic responsiveness to i.t. morphine (i.e., tolerance) following daily i.t. application of 15 µg morphine for 6 days, n=6. I.t. CSD+PP2 (10 µg and 300 ng, respectively) pre-treatment (45 min before i.t. morphine) completely prevented tolerance development (filled circles; n=10). Vehicle (DMSO) for CSD+PP2, which was included in the control group had no effect on tolerance development. Panel B: Dose-responsiveness to i.t. morphine was determined in morphine naive rats (opened circle; n=7), morphine tolerant rats (filled circle; n=6), and rats that had received i.t. CSD+PP2 45 min prior to chronic daily i.t. morphine (CM) (filled square; n=10). Morphine tolerant rats required 3.4 times more morphine than naive rats to elicit comparable anti-allodynia (ED50=13.1 µg vs. 3.9 µg). In contrast, i.t. morphine was essentially equi-effective in CSD+PP2+CM group vs. opioid naïve rats, i.e., CSD+PP2 eliminated morphine anti-allodynic tolerance. Panel C: Chronic i.t. CSD+PP2 daily for 6 days did not significantly affect either basal von Frey thresholds or i.t. morphine dose responsiveness, in the absence of chronic i.t. morphine treatment. n=4 for each point. CSD (caveolin-1 scaffolding domain peptide); PP2 (selective Src inhibitor); CM (chronic morphine).
Figure 1: I.t. CSD and PP2 block tolerance development to i.t. morphine. Panel A: Opened circle (control group) illustrates the complete loss of anti-allodynic responsiveness to i.t. morphine (i.e., tolerance) following daily i.t. application of 15 µg morphine for 6 days, n=6. I.t. CSD+PP2 (10 µg and 300 ng, respectively) pre-treatment (45 min before i.t. morphine) completely prevented tolerance development (filled circles; n=10). Vehicle (DMSO) for CSD+PP2, which was included in the control group had no effect on tolerance development. Panel B: Dose-responsiveness to i.t. morphine was determined in morphine naive rats (opened circle; n=7), morphine tolerant rats (filled circle; n=6), and rats that had received i.t. CSD+PP2 45 min prior to chronic daily i.t. morphine (CM) (filled square; n=10). Morphine tolerant rats required 3.4 times more morphine than naive rats to elicit comparable anti-allodynia (ED50=13.1 µg vs. 3.9 µg). In contrast, i.t. morphine was essentially equi-effective in CSD+PP2+CM group vs. opioid naïve rats, i.e., CSD+PP2 eliminated morphine anti-allodynic tolerance. Panel C: Chronic i.t. CSD+PP2 daily for 6 days did not significantly affect either basal von Frey thresholds or i.t. morphine dose responsiveness, in the absence of chronic i.t. morphine treatment. n=4 for each point. CSD (caveolin-1 scaffolding domain peptide); PP2 (selective Src inhibitor); CM (chronic morphine).
I.t. treatment with CSD+PP2 reverses spinal opioid anti-allodynic tolerance. Prevention of the development of spinal opioid anti-allodynic tolerance by i.t. CSD+PP2 does not necessarily indicate the ability of this treatment to reverse tolerance, once it has fully developed. In order to determine if i.t. CSD+PP2 was also able to acutely reverse opioid anti-allodynic tolerance, we determined the effect of intrathecally applying CSD+PP2 on i.t. morphine anti-allodynia subsequent to tolerance development (Figure 2A). One-way ANOVA revealed a significant treatment effect among days 1, 9, and 11 (F2,21=58.4, p < 0.0001), significant differences being observed between effects on day 1 vs. day 9 (when anti-allodynic tolerance was fully manifest) (P < 0.001) and effects on day 9 vs. day 11 (when i.t. CSD+PP2 resulted in the biggest reversal) (p < 0.01), but not between days 1 and 11 (p>0.05). The latter comparison revealed that tolerance reversal was complete. As illustrated in Figure 2A (n=8), following 9-days of spinal morphine treatment, morphine tolerance was fully manifest, essentially eliminating the totality of morphine’s anti-allodynic effect. The i.t. application of CSD+PP2 on day 10 (45 min prior to intrathecally administering morphine) restored >95% of the initial i.t. morphine-induced anti-allodynia.
We also investigated the effect of i.t. CSD+PP2 on tail flick latency (Figure 2B) in order to investigate whether or not the acute anti-tolerance effect of i.t. CSD+PP2 was pain modality specific. As was observed for neuropathetic pain, one-way ANOVA revealed a significant treatment effect (F2,11=17.5, p < 0.001; n=5) among days 1, 9, and 11, significant differences being observed on day 1 vs. day 9 (when tolerance was fully manifest) (P<0.001) and on day 9 vs. 11 (when CSD+PP2 showed the biggest reversal) (p < 0.01). No treatment effect was observed between day 1 and day 11 (p>0.05), indicating that tolerance reversal was essentially complete, i.t. CSD+PP2 restoring >90% of the analgesic effect of i.t. morphine manifest prior to chronic spinal morphine (Figure 2).
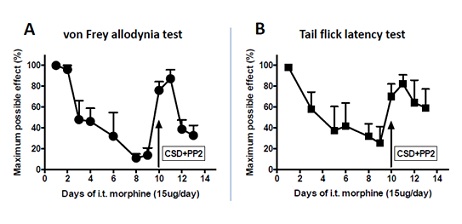 Figure 2: I.t. treatment with CSD and PP2 reverses i.t. morphine tolerance. Panel A: The von Frey allodynia test showed that i.t. morphine had lost most of its anti-allodynic effectiveness after 9 days of daily i.t. morphine treatment (15 µg). I.t. CSD+PP2 (10 µg and 300 ng, respectively) applied on day 10, 45 min before i.t. morphine, fully restored spinal anti-allodynic morphine responsiveness, which lasted for 2 days (n=8). Panel B: The tail flick latency test was also used to assess i.t. morphine analgesic tolerance. As is illustrated in Panel A, i.t. morphine lost most of its analgesic effect 9 days following daily 15 µg i.t. morphine treatment. I.t. CSD+PP2, applied on day 10, 45 min before i.t. morphine, fully restored morphine antinociception, which lasted for 4 days (n=5). CSD (Caveolin-1 Scaffolding Domain peptide); PP2 (selective Src inhibitor).
Figure 2: I.t. treatment with CSD and PP2 reverses i.t. morphine tolerance. Panel A: The von Frey allodynia test showed that i.t. morphine had lost most of its anti-allodynic effectiveness after 9 days of daily i.t. morphine treatment (15 µg). I.t. CSD+PP2 (10 µg and 300 ng, respectively) applied on day 10, 45 min before i.t. morphine, fully restored spinal anti-allodynic morphine responsiveness, which lasted for 2 days (n=8). Panel B: The tail flick latency test was also used to assess i.t. morphine analgesic tolerance. As is illustrated in Panel A, i.t. morphine lost most of its analgesic effect 9 days following daily 15 µg i.t. morphine treatment. I.t. CSD+PP2, applied on day 10, 45 min before i.t. morphine, fully restored morphine antinociception, which lasted for 4 days (n=5). CSD (Caveolin-1 Scaffolding Domain peptide); PP2 (selective Src inhibitor).
I.t. CSD and PP2, each administered individually, blocks the development as well as maintenance of spinal opioid anti-allodynic tolerance induced by chronic i.t. morphine. We hypothesized that CSD and PP2 would interfere with tolerance by acting at two sequentially interconnected components of tolerance mechanisms, CSD interfering with scaffolding by competing with the scaffolding domain within Cav1 while PP2 inhibits Src kinase, known to phosphorylate Tyr 114 of Cav1, thereby modulating its scaffolding ability. Accordingly, we tested the hypothesis that the individual i.t. application of either CSD (10 µg; Figure 3A) or PP2 (300 ng; Figure 3B) would be sufficient to interrupt opioid anti-allodynic tolerance. For the CSD treated group (n=6), one-way ANOVA showed a significant treatment effect (F2,15=14.5, p < 0.001) between day 1 and day 7 (when tolerance was fully manifest) (P < 0.001) as well as between day 7 and day 8 (when CSD showed the biggest reversal) (p < 0.01), but not between day 1 and day 8 (p>0.05). The latter comparison reveals that tolerance reversal was complete.
The PP2 treated group followed suit. One-way ANOVA showed a significant treatment effect (F2,12=14.3, p < 0.001) between day 1 and day 8 (when tolerance was fully manifest) (P < 0.001) as well as between day 8 and day 9 (when PP2 produced the biggest reversal) (p < 0.05) (n=5). As is shown in figure 3, by day 7 of spinal morphine treatment, tolerance had developed to i.t. morphine, essentially eliminating the totality of its anti-allodynic effects. On day 9, i.t. PP2 (Figure 3B), applied intrathecally 45 min prior to the i.t. morphine, attenuated spinal opioid anti-allodynic tolerance, substantially restoring the ability of i.t. morphine to elevate allodynic (von Frey) thresholds.
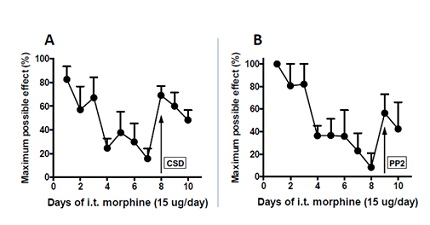 Figure 3: I.t. treatment with either CSD or PP2 reverses i.t. morphine tolerance. Spinal anti-allodynic tolerance to i.t. morphine was generated as described in the legend to figure 1. The von Frey allodynia test revealed that i.t. morphine had lost most of its anti-allodynic effect 7-8 days following daily i.t. 15 µg morphine. CSD (10 µg) (Panel A) or PP2 (300 ng) (Panel B) was intrathecally applied 45 min before i.t. morphine on day 8 or 9 of chronic morphine treatment when maximal morphine tolerance was manifest. I.t. CSD fully restored i.t morphine antinociception, whereas i.t. PP2 restored approximately 60% of opioid naïve i.t. morphine responsiveness. The reversal of morphine tolerance by either CSD or PP2 lasted for multiple days. n=6 and 5 for CSD and PP2, respectively. CSD (Caveolin-1 Scaffolding Domain peptide); PP2 (selective Src inhibitor).
Figure 3: I.t. treatment with either CSD or PP2 reverses i.t. morphine tolerance. Spinal anti-allodynic tolerance to i.t. morphine was generated as described in the legend to figure 1. The von Frey allodynia test revealed that i.t. morphine had lost most of its anti-allodynic effect 7-8 days following daily i.t. 15 µg morphine. CSD (10 µg) (Panel A) or PP2 (300 ng) (Panel B) was intrathecally applied 45 min before i.t. morphine on day 8 or 9 of chronic morphine treatment when maximal morphine tolerance was manifest. I.t. CSD fully restored i.t morphine antinociception, whereas i.t. PP2 restored approximately 60% of opioid naïve i.t. morphine responsiveness. The reversal of morphine tolerance by either CSD or PP2 lasted for multiple days. n=6 and 5 for CSD and PP2, respectively. CSD (Caveolin-1 Scaffolding Domain peptide); PP2 (selective Src inhibitor).
I.t. CSD blocks both the development and maintenance of spinal opioid tolerance induced by chronic systemic morphine. Recognizing that opioids are frequently given systemically, in addition to intrathecally, we investigated the ability of i.t. CSD to prevent and/or reverse spinal opioid anti-allodynic tolerance induced by systemic chronic morphine, administered via the s.c. implantation of morphine base pellets. These data are shown in Figure 4. Figure 4A included 3 groups: animals receiving s.c. morphine pellets (n=6), morphine-pelleted animals receiving i.t. CSD pretreatment, administered 45 min before morphine pellet implantation on days 1, 3 and 5 (n=7) and morphine-pelleted animals receiving i.t. pretreatment with scrambled CSD (S-CSD; administered as described for i.t. CSD) (n=7). Two-way ANOVA showed a significant treatment effect among the 3 groups (F2,51=153.4, p < 0.0001), significant differences being observed between the CSD and S-CSD pretreated groups (p < 0.001) as well as between chronic morphine pelleted rats with vs. without i.t. CSD pretreatment p (p < 0.0001). Two-days after s.c. implantation of morphine base pellets in the absence of i.t. CSD, the anti-allodynic effect of the systemic morphine was substantially reduced relative to day 1, when cutoff von Frey thresholds were manifest (data not shown). In fact, von Frey thresholds continued to decline through day 8. Notably, in the presence of i.t. CSD (10 µg), the anti-allodynia produced by the implanted morphine pellets was fully manifest, as reflected by the von Frey thresholds reaching cutoff. Blockade of opioid anti-allodynic tolerance development persisted through day 8, 3 days after the last i.t. application of CSD. On day 9 (4 days after the last i.t. application of CSD), when spinal CSD concentrations presumably started to decline, the manifestation of opioid anti-allodynic tolerance began to re-emerge, indicating that blockade of tolerance development by CSD was not an artifact of the random occurrence of tolerance resistance in this particular group of rats. In contrast to CSD, i.t. pretreatment with S-CSD (10 µg applied 45 min before i.t. morphine) failed to alter morphine tolerance development, i.e., there was no difference in tolerance development to chronic systemic morphine between animals that received i.t. S-CSD vs. no spinal treatment.
Spinal treatment with CSD was also effective in reversing systemic morphine-induced spinal opioid tolerance, once it had been fully developed (Figure 4B). One-way ANOVA showed a significant treatment effect (F2,24=6.03, p<0.01, n=8) between day 1 and day 7 (when tolerance was fully manifest) (P < 0.01) as well as between day 7 and day 8 (indicating acute tolerance reversal by CSD) (p < 0.05), but not between day 1 and day 8 (p>0.05). The latter comparison indicating that tolerance had been acutely eliminated. Figure 4B illustrates that chronic systemic morphine had lost virtually all of its anti-allodynic effect seven days following the onset of pelleting. Forty-five min after 10 µg i.t. CSD, spinal morphine analgesic tolerance was essentially fully mitigated, i.t. morphine producing a robust anti-allodynia that was comparable to that produced prior to the onset of morphine pelleting, i.e., i.t. CSD fully restored i.t. morphine anti-allodynic action. Full reversal of tolerance development was also effectuated after i.t. application of lower dose of CSD (3, 1, or 0.33 µg; data not shown). The opioid tolerance-reversing effect of i.t. CSD persisted in excess of 24 hours. In contrast, i.t. S-CSD had no effect on the chronic systemic morphine-induced tolerance (p>0.05 compare to day 7, n=6).
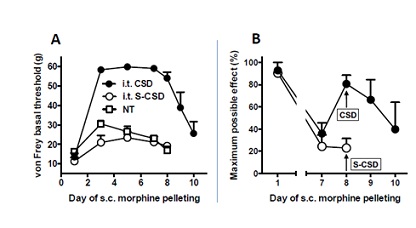 Figure 4: I.t. CSD, alone, prevents as well as reverses spinal morphine anti-allodynic tolerance induced by systemic morphine. Panel A: von Frey thresholds of the rear paw ipsilateral to Spinal Nerve Ligation (SpNL) were quantified immediately prior to every morphine pellet implantation (performed on day 1, 3 and 5) and/or other daily i.t. treatments. CSD or S-CSD) was intrathecally applied 45 min before s.c. morphine pellet implantation. Day 1 represents opioid naïve von Frey thresholds. Throughout the ensuing week, the anti-allodynic effects of the onboard s.c. morphine pellets were robustly manifest in morphine-pelleted rats that had received i.t. CSD. This was reflected by the increase to cutoff in von Frey basal thresholds, which persisted for 3-days following the last i.t. CSD treatment. In contrast, the anti-allodynia resulting from the s.c. morphine pellets remained minimal in rats that had received i.t. S-CSD (open circle) or no i.t. treatment (open square). N=7, 7 and 6, for CSD, S-CSD and no i.t. treatment group, respectively. Panel B: I.t. CSD or S-CSD (10 µg) was applied 45 min prior to i.t. morphine on day 8 when morphine anti-allodynic tolerance was fully manifest. CSD essentially fully restored i.t. morphine anti-allodynic action (n=8). This anti-allodynic action lasted for more than 24 hours. In contrast, i.t. S-CSD failed to alter i.t. morphine tolerance (n=6). CSD (Caveolin-1 Scaffolding Domain peptide); S-CSD (Scrambled Caveolin-1 Scaffolding Domain peptide); NT (no treatment); SpNL (Spinal Nerve Ligation).
Figure 4: I.t. CSD, alone, prevents as well as reverses spinal morphine anti-allodynic tolerance induced by systemic morphine. Panel A: von Frey thresholds of the rear paw ipsilateral to Spinal Nerve Ligation (SpNL) were quantified immediately prior to every morphine pellet implantation (performed on day 1, 3 and 5) and/or other daily i.t. treatments. CSD or S-CSD) was intrathecally applied 45 min before s.c. morphine pellet implantation. Day 1 represents opioid naïve von Frey thresholds. Throughout the ensuing week, the anti-allodynic effects of the onboard s.c. morphine pellets were robustly manifest in morphine-pelleted rats that had received i.t. CSD. This was reflected by the increase to cutoff in von Frey basal thresholds, which persisted for 3-days following the last i.t. CSD treatment. In contrast, the anti-allodynia resulting from the s.c. morphine pellets remained minimal in rats that had received i.t. S-CSD (open circle) or no i.t. treatment (open square). N=7, 7 and 6, for CSD, S-CSD and no i.t. treatment group, respectively. Panel B: I.t. CSD or S-CSD (10 µg) was applied 45 min prior to i.t. morphine on day 8 when morphine anti-allodynic tolerance was fully manifest. CSD essentially fully restored i.t. morphine anti-allodynic action (n=8). This anti-allodynic action lasted for more than 24 hours. In contrast, i.t. S-CSD failed to alter i.t. morphine tolerance (n=6). CSD (Caveolin-1 Scaffolding Domain peptide); S-CSD (Scrambled Caveolin-1 Scaffolding Domain peptide); NT (no treatment); SpNL (Spinal Nerve Ligation).
I.t. CSD+PP2 restores i.t. morphine dose responsiveness in animals treated chronically with systemic morphine. In order to assess the full extent of the ability of i.t. CSD+PP2 to reverse tolerance to spinal morphine’s anti-allodynic actions in animals receiving chronic systemic morphine, we investigated the ability of i.t. CSD+PP2 to restore spinal morphine anti-allodynic dose responsiveness (Figure 5). Groups consisted of opioid naïve animals (‘before pelleting’), opioid tolerant animals (‘after pelleting’) and morphine pelleted animals that also received a one-time i.t. application of CSD+PP2) (‘pelleting, CSD+PP2’), n=8 for each group. Two-way ANOVA revealed significant treatment effects among the groups (F2,47=194.0, p<0.0001). As expected, i.t. morphine dose responsiveness obtained in opioid naïve animals was significantly different from i.t. dose responsiveness obtained in the morphine pelleted animals (p<0.01). Strikingly, i.t. morphine anti-allodynic dose responsiveness obtained in morphine pelleted (opioid tolerant) animals also significantly differed from that obtained in morphine pelleted animals that had received a one-time i.t. application of CSD+PP2, 45 min prior to quantifying i.t. morphine dose responsiveness (p < 0.001). Moreover, morphine dose responsiveness obtained in the latter group did not significantly differ from that obtained in opioid naïve animals (p>0.05), indicating that i.t. CSD+PP2 restored normative spinal morphine anti-allodynia. These data are shown in figure 5, which illustrates that chronic systemic morphine shifted i.t. morphine ED50 from 5.5 µg to 19.7 µg. The totality of this rightward shift in morphine dose responsiveness (anti-allodynic tolerance) was obliterated following the i.t. 45 min pretreatment with CSD+PP2 (10 µg and 300 ng, respectively), i.e., i.t. CSD+PP2 pretreatment of morphine tolerant animals reinstated spinal morphine anti-allodynic responsiveness (ED50=6.4 µg), which was indistinguishable from that observed in opioid naïve rats (5.5 µg).
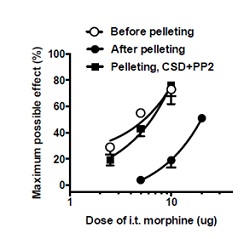 Figure 5: I.t. CSD and PP2 reverse spinal tolerance to systemic morphine. Dose-responsiveness to i.t. morphine in opioid naive rats (opened circle; n=8), rats receiving chronic systemic morphine via s.c. implanted morphine pellets (filled circle; n=8) and rats receiving chronic systemic morphine along with a one-time i.t. application of CSD+PP2 (10 µg and 300 ng, respectively; administered 45 min prior to assessing i.t. morphine dose-responsiveness) (filled square; n=8). As expected, the i.t. morphine ED50 was 3.6-fold greater in chronic systemic morphine-treated (tolerant) rats than opioid naive rats (19.7 µg vs. 5.5 µg), reflecting anti-allodynic tolerance development. In contrast, the i.t. morphine ED50 observed in chronic morphine-treated rats that received i.t. CSD+PP2 prior to dose-response quantification was essentially the same as that of opioid naïve rats (6.4 µg vs. 5.5 µg), underscoring the ability of i.t. CSD+PP2 to acutely eliminate morphine tolerance, even after its full development. CSD (Caveolin-1 Scaffolding Domain peptide); PP2 (selective Src inhibitor).
Figure 5: I.t. CSD and PP2 reverse spinal tolerance to systemic morphine. Dose-responsiveness to i.t. morphine in opioid naive rats (opened circle; n=8), rats receiving chronic systemic morphine via s.c. implanted morphine pellets (filled circle; n=8) and rats receiving chronic systemic morphine along with a one-time i.t. application of CSD+PP2 (10 µg and 300 ng, respectively; administered 45 min prior to assessing i.t. morphine dose-responsiveness) (filled square; n=8). As expected, the i.t. morphine ED50 was 3.6-fold greater in chronic systemic morphine-treated (tolerant) rats than opioid naive rats (19.7 µg vs. 5.5 µg), reflecting anti-allodynic tolerance development. In contrast, the i.t. morphine ED50 observed in chronic morphine-treated rats that received i.t. CSD+PP2 prior to dose-response quantification was essentially the same as that of opioid naïve rats (6.4 µg vs. 5.5 µg), underscoring the ability of i.t. CSD+PP2 to acutely eliminate morphine tolerance, even after its full development. CSD (Caveolin-1 Scaffolding Domain peptide); PP2 (selective Src inhibitor).
I.t. PP2 reverses spinal opioid tolerance induced by systemic morphine. As we observed for CSD, i.t. PP2 (300 ng applied on day 8, 45 min before i.t. morphine), fully restored i.t. morphine anti-allodynic effects, which had been diminished by approximately 80% (Figure 6). One-way ANOVA showed a significant treatment effect (F2,18=301.7, p<0.0001). The significant effect was between day 1 and day 7 (when tolerance fully manifested) (P<0.0001) as well as between day 7 and day 8 (when CSD showed biggest reversal) (p<0.0001). In contrast, a significant effect was not observed between day 1 and day 8 (p>0.05; n=7), indicating that i.t. PP2 fully reversed spinal morphine tolerance. Interestingly, on day 9, one day after i.t. PP2, opioid anti-allodynic tolerance was once again manifest, indicating that the reversal observed on day 8 did not result from a random, sudden loss of tolerance, confounding interpretation.
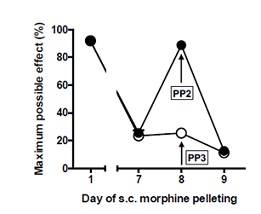 Figure 6: I.t. PP2 reverses spinal tolerance induced by systemic morphine. Experiments were performed as described in the legend to Fig. 5. I.t. PP2 (300 ng; applied on day 8, 45 min before i.t. morphine) essentially fully restored i.t. morphine anti-allodynic action. This effect lasted less than 24 hours. In contrast, i.t. PP3 (300 ng), a negative control for PP2, applied 45 min before i.t. morphine, failed to alter anti-allodynic action of i.t. morphine. These results indicate that i.t. PP2 can reverse spinal morphine anti-allodynic tolerance induced by systemic morphine (as was observed for i.t. CSD). PP2 (selective Src inhibitor); PP3 (negative control for PP2).
Figure 6: I.t. PP2 reverses spinal tolerance induced by systemic morphine. Experiments were performed as described in the legend to Fig. 5. I.t. PP2 (300 ng; applied on day 8, 45 min before i.t. morphine) essentially fully restored i.t. morphine anti-allodynic action. This effect lasted less than 24 hours. In contrast, i.t. PP3 (300 ng), a negative control for PP2, applied 45 min before i.t. morphine, failed to alter anti-allodynic action of i.t. morphine. These results indicate that i.t. PP2 can reverse spinal morphine anti-allodynic tolerance induced by systemic morphine (as was observed for i.t. CSD). PP2 (selective Src inhibitor); PP3 (negative control for PP2).
Importantly, the negative control for PP2, PP3, was devoid of any effect on opioid anti-allodynic tolerance. One-way ANOVA of the PP3 group showed a significant treatment effect (F2,6=185.6, p < 0.0001), reflecting spinal morphine anti-allodynic tolerance. The significant effect was between day 1 and day 7 (when tolerance was fully manifested) (P < 0.0001) as well as between day 1 and day 8 (P < 0.0001), illustrating the persistence of this tolerance after i.t. PP3, but not between day 7 and day 8 (p>0.05). The latter underscores the inability of i.t. PP3 to reverse tolerance. n=3.
Discussion
The development of opioid analgesic tolerance results in the requirement for profound dose escalation. Since this exacerbates side effects and addiction liability, the need for dose escalation has long been a major confound in the clinical utilization of opioids for managing nociception. Current findings reveal that the scaffolding protein Cav1 and the nonreceptor tyrosine kinase Src (which phosphorylates Cav1, thereby altering its scaffolding properties) are two likely targets for developing novel adjunctive pharmacotherapies to mitigate, if not abolish, opioid analgesic tolerance.
The hypothesized relevance of protein scaffolding to the development of opioid tolerance was predicated on multiple factors. These include (a) the presence of MOR (and the kappa-opioid receptor) within multimeric signaling complexes [56,103], which invariably require a structural backbone for their formation; (b) the ability of chronic morphine to induce coordinated, interrelated changes in a wide spectrum of signaling molecules downstream from MOR, which is likely to be facilitated by protein scaffolding; as would be (c) the increased emergence and prominence of new signaling strategies in response to chronic morphine, well documented to occur in response to chronic morphine [35,44].
As hypothesized, the combined i.t. application of CSD (which binds to the scaffolding domain of Cav1, thereby effectively blocking scaffolding) and PP2 (which inhibits Src phosphorylation of Cav1) eliminated the development of spinal tolerance to the anti-allodynic effect of intrathecally-applied morphine. Importantly, i.t. CSD+PP2 abolished tolerance development irrespective of the route of chronic morphine administration, abolishing tolerance induced by chronic morphine applied spinally (via daily i.t. application) as well as systemically (via s.c. implantation of morphine base pellets). Since in our formulation, Src is posited to act upstream of Cav1, modifying its scaffolding functionality via phosphorylation, we anticipated that the individual i.t. application of CSD (i.e., interrupting Cav1 scaffolding) or PP2 (inhibiting Cav1 phosphorylation) would mitigate tolerance development to the same extent as would applying CSD together with PP2. As expected, i.t. CSD, in the absence of PP2, or PP2 (in the absence of CSD) retained their ability to eliminate spinal morphine anti-allodynic tolerance. This underscores the putative clinical utility of interfering with Cav1 scaffolding as an adjunctive pharmacotherapy accompanying the use of opioids for pain management.
Many of the proposed biochemical underpinnings of opioid tolerance are not consonant with acute reversal of tolerance once it has fully developed. For example, the long-held view that MOR internalization/downregulation and adenylyl cyclase super activation are foundational to opioid analgesic tolerance [104-106] would not be expected to be readily reversed within min following pharmacological intervention, thereby restoring naïve levels of anti-allodynic responsiveness to i.t. morphine. Strikingly, this is exactly what was observed following i.t. CSD+PP2 or their individual i.t. application. In fact, i.t. CSD (0.33 µg) unmasked robust morphine anti-allodynia (from on boarded morphine pellets) in systemic morphine tolerant animals, even in the absence of virtually any pretreatment with CSD. Of note, the acute reversal of morphine anti-allodynic tolerance by CSD (but not S-CSD) and/or PP2 (but not PP3) was not permanent. Within 24-72 hrs, tolerance was once again observed. This reinstatement of tolerance presumably resulted from the run-down of CSD or PP2 concentrations, resulting in the reinstatement of Cav1 scaffolding functionality, characteristic of the opioid tolerant condition. The acute reversal and reestablishment of morphine anti-allodynic tolerance reveals a fluid, temporally dynamic dimension of opioid tolerance, not explicitly provided for in current models of tolerance. Indeed, it is becoming increasingly appreciated that protein function is critically dependent on the milieu of other proteins and other molecules that interact with each other to control a signaling process. The dynamics of these protein communities was originally shown to regulate metabolic processes [107-110], which itself can influence cellular signal transduction at multiple points, e.g., membrane localization of growth factor receptors [111], allowing for adaptation and rapid changes based on cellular and environmental stimuli. The role of metabolomic supramolecular complexes, transient assemblies of proteins that form to regulate seminal processes and the temporal, spatial and conditional dimensions thereof, in modulating neuronal functional states remains unknown. One can envision that just like metabolomes are highly dynamic assemblies of proteins, the effects of their disassembly on the neuronal tolerant state can be equally temporally dynamic.
The magnitude of acute reversal by i.t. CSD and/or PP2 of spinal morphine anti-allodynic tolerance as reflected by unmasking the anti-allodynic effects of on boarded morphine pellets were quite variable. This contrasts with the magnitude of the acute reversal by i.t. CSD and/or PP2 of tolerance to the anti-allodynic effects of intrathecally-applied morphine, which is consistently robust. This dichotomy likely results from the spinally restricted loci of action of intrathecally applied CSD and/or PP2 since such localization would more consistently impact tolerance to the anti-allodynia mediated by spinal opioid receptors (vis a vis i.t. morphine) than the anti-allodynia mediated by opioid receptors located supraspinally as well as spinally, as occurs with systemic morphine. The ability of CSD and/or PP2 to prevent and/or reverse supraspinal morphine anti-allodynic tolerance when they are applied to supraspinal sites remains to be determined.
The variable ability of i.t. CSD/PP2 to unmask the anti-allodynic effects of subcutaneously implanted morphine pellets also contrasts with our observation that the identical i.t. treatment prior to implanting morphine base pellets invariably and robustly maintained the anti-allodynic effects of implanted morphine pellets (i.e., prevented tolerance development) such that the von Frey cutoff threshold was routinely manifest. This dichotomy can result from the bidirectional communication between the spinal cord and brain, e.g., altered spinal cord excitability alters ascending messages, which in turn influences descending modulation arising from the midbrain (e.g., periaqueductal gray) and brainstem (e.g., rostroventromedial medulla) [112,113]. A major difference between preventing vs. acutely reversing spinal opioid tolerance that results from chronic systemic morphine is that spinal tolerance prevention would obviate any influence of spinal opioid tolerance on ascending spinal brain communication (e.g. attenuating endogenous opioid modulation of ascending spinal nociceptive pathways) and resulting supraspinal adaptations, whereas acutely reversing spinal tolerance subsequent to its development would not. Moreover, the temporal profile for the offset of these adaptations might not coincide with that of the acute reversal of spinal opioid tolerance (and presumably the reinstatement of the unfettered activity of spinal ascending pathways). These considerations imply that while prevention and reversal of morphine anti-allodynic tolerance by i.t. CSD/PP2 result from interrupting a common mechanism (Cav1 scaffolding), they likely do not share identical biochemical sequelae.
Multiple clinical reports indicate that persistent opioid exposure can, paradoxically, result in allodynia and hyperalgesia, which often differs from the original pain that is being treated [114-117]. Analogous observations have also been reported in laboratory animals [118]. Interestingly, supraspinal adaptations to chronic systemic morphine (e.g., in the Rostroventromedial Medulla (RVM) and dorsolateral funiculus) has been shown to mediate chronic opioid-induced nociception (i.e., hyperalgesia, allodynia) as well as spinal morphine antinociceptive tolerance [119]. In fact, the former has been causally associated with the latter [119]. This is noteworthy since i.t. CSD and/or PP2 similarly prevented/reversed spinal morphine anti-allodynic tolerance produced by chronic morphine delivered either intrathecally or systemically, indicating that CSD and or PP2 interfere with a fundamental, shared component of multiple tolerance-producing adaptations. Additionally, these considerations suggest that CSD and/or PP2 could be effective in blocking/reversing chronic opioid-induced allodynia/hyperalgesia, further extending the clinical utility of opioids in chronic pain management.
Whereas CSD is specific for interrupting Cav1 scaffolding, Src has many substrates that could influence the development and maintenance of opioid tolerance. For example, chronic morphine-induced activation of Src can not only result in increased tyrosine phosphorylation of Cav1, altering its scaffolding properties, but also the tyrosine phosphorylation of MOR, (Kramer et al.,; Zhang, et al.,), which has been causally linked to the chronic morphine-induced activation of AC (AC super activation) as well as the conversion of MOR from a classical G-protein coupled receptor to a receptor tyrosine kinase-like receptor [46]. Additionally, Src has been reported to participate in delta opioid receptor activation of ERK [120]. Src activation by chronic opioids and consequent phosphorylation of multiple signaling molecules could concomitantly activate parallel tolerant mechanisms, all of which might be eliminated by Src inhibition. While this, hypothetically, could account for the ability of PP2 to interrupt opioid tolerance, it would not explain the ability of CSD to do the same, as was currently found. The most parsimonious explanation for our finding that PP2 and CSD have concordant effects on opioid anti-allodynic tolerance would be that PP2 and CSD acted at sequential steps in a common mechanism, PP2 inhibiting Src phosphorylation of Cav1, preventing the altered scaffolding that is critical to anti-allodynic morphine tolerance, CSD eliminating scaffolding by binding to the scaffolding site within Cav1.
Many cellular adaptations to chronic morphine have been described in cells maintained in culture and laboratory animal models. It remains unclear whether the lack of clinical utility of the most, if not all, of the previously intimated opioid tolerance mitigating therapies resulted from failure of the test or failure to test. Regardless, theoretically, there is a much greater likelihood for clinical success of developing opioid anti-tolerance pharmacotherapies that disrupt Cav1 scaffolding functionality in the presence of chronic opioids since doing so has the potential to concomitantly interrupt multiple analgesic/anti-allodynic tolerance mechanisms. The patent (WO 2021/158317 A1) processing has been delaying the submission for publication of this work, which provides an opportunity for other group to publish closely related work, yet with different focus and underlying mechanisms [86]. Although it tested the effect of Cav1 on morphine-induced hyperalgesia, it supports and emphasizes the clinical potential of our findings in preventing as well as reversing opioid tolerance in treating severe pain.
Disclosure Statement
The authors report there are no competing interests to declare.
Funding Details
This work was supported by the NIDA under Grant R01DA043774.
References
- Kinnunen M, Piirainen P, Kokki H, Lammi P, Kokki M (2019) Updated Clinical Pharmacokinetics and Pharmacodynamics of Oxycodone. Clin Pharmacokinet 58: 705-725.
- Rapp SE, Ready BL, Nessly ML (1995) Acute pain management in patients with prior opioid consumption: A case-controlled retrospective review. Pain 61: 195-201.
- McQuay H (1999) Opioids in pain management. Lancet 353: 2229-2232.
- McQuay HJ, Bullingham RE, Moore RA (1981) Acute opiate tolerance in man. Life Sci 28: 2513-2517.
- Houde RW (1985) The analgesic connection: the Nathan B Eddy memorial lecture, in Problems of drug dependence. NIDA Res Monogr: 4-13.
- Houde RW, Wallenstein SL, Beaver WT (1966) Evaluation of analgesics in patients with cancer pain, in International encyclopedia of pharmacology and therapeutics. Pergamon Press, Oxford, Oxford, UK.
- Buntin-Mushock C, Phillip L, Moriyama K, Palmer PP (2005) Age-dependent opioid escalation in chronic pain patients. Anesth Analg 100: 1740-1745.
- Nicholson B, Verma S (2004) Comorbidities in chronic neuropathic pain. Pain Med 5: 9-27.
- Baliki MN, Geha PY, Apkarian AV, Chialvo DR (2008) Beyond feeling: Chronic pain hurts the brain, disrupting the default-mode network dynamics. J Neurosci 28: 1398-1403.
- Rodriguez-Raecke R, Niemeier A, Ihle K, Ruether W, May A (2009) Brain gray matter decrease in chronic pain is the consequence and not the cause of pain. J Neurosci 29: 13746-13750.
- Apkarian AV, Sosa Y, Krauss BR, Thomas PS, Fredrickson BE, et al. (2004) Chronic pain patients are impaired on an emotional decision-making task. Pain 108: 129-136.
- Apkarian AV, Sosa Y, Sonty S, Levy RM, Harden RN, et al. (2004) Chronic back pain is associated with decreased prefrontal and thalamic gray matter density. J Neurosci 24: 10410-10415.
- Schwartz N, Temkin P, Jurado S, Lim BK, Heifets BD, et al. (2014) Chronic pain. Decreased motivation during chronic pain requires long-term depression in the nucleus accumbens. Science 345: 535-542.
- Ljungqvist O, Scott M, Fearon KC (2017) Enhanced Recovery After Surgery: A Review. JAMA Surg 152: 292-298.
- Chavkin C, Goldstein A (1984) Opioid receptor reserve in normal and morphine-tolerant guinea pig ileum myenteric plexus. Proc Natl Acad Sci USA 81: 7253-7257.
- Cox BM, Crowder AT (2004) Receptor domains regulating mu opioid receptor uncoupling and internalization: Relevance to opioid tolerance. Mol Pharmacol 65: 492-495.
- Chakrabarti S, Law PY, Loh HH (1995) Neuroblastoma Neuro2A cells stably expressing a cloned mu-opioid receptor: A specific cellular model to study acute and chronic effects of morphine. Brain Res Mol Brain Res 30: 269-278.
- Sim LJ, Selley DE, Dworkin SI, Childers SR (1996) Effects of chronic morphine administration on mu opioid receptor-Stimulated [35S]GTPgammaS autoradiography in rat brain. J Neurosci 16: 2684-2692.
- Patel MB, Patel CN, Rajashekara V, Yoburn BC (2002) Opioid agonists differentially regulate mu-opioid receptors and trafficking proteins in vivo. Mol Pharmacol 62: 1464-1470.
- Yoburn BC, Billings B, Duttaroy A (1993) Opioid receptor regulation in mice. J Pharmacol Exp Ther 265: 314-320.
- Shen J, Benedict Gomes A, Gallagher A, Stafford K, Yoburn BC (2000) Role of cAMP-dependent protein kinase (PKA) in opioid agonist-induced mu-opioid receptor downregulation and tolerance in mice. Synapse 38: 322-327.
- Trafton JA, Basbaum AI (2004) [d-Ala2,N-MePhe4,Gly-ol5]enkephalin-induced internalization of the micro opioid receptor in the spinal cord of morphine tolerant rats. Neuroscience 125: 541-543.
- Madia PA, Navani DM, Yoburn BC (2012) [(35)S]GTPγS binding and opioid tolerance and efficacy in mouse spinal cord. Pharmacol Biochem Behav 101: 155-165.
- Grecksch G, Just S, Pierstorff C, Imhof AK, Glück L, et al. (2011) Analgesic tolerance to high-efficacy agonists but not to morphine is diminished in phosphorylation-deficient S375A μ-opioid receptor knock-in mice. J Neurosci 31: 13890-13896.
- Walwyn W, Evans CJ, Hales TG (2007) Beta-arrestin2 and c-Src regulate the constitutive activity and recycling of mu opioid receptors in dorsal root ganglion neurons. J Neurosci 27: 5092-5104.
- Brady LS, Herkenham M, Long JB, Rothman RB (1989) Chronic morphine increases mu-opiate receptor binding in rat brain: A quantitative autoradiographic study. Brain Res 477: 382-386.
- Rothman RB, Yang HY, Long JB (1990) Upregulation of rat brain opioid receptors by the chronic administration of morphine: Possible evidence for an anti-opiate model of tolerance and dependence. NIDA Res Monogr 105: 264-270.
- Chakrabarti S, Madia PA, Gintzler AR (2016) Selective up-regulation of functional mu-opioid receptor splice variants by chronic opioids. J Neurochem 136: 1119-1130.
- Sim-Selley LJ, Selley DE, Vogt LJ, Childers SR, Martin TJ (2000) Chronic heroin self-administration desensitizes mu opioid receptor-activated G-proteins in specific regions of rat brain. J Neurosci 20: 4555-4562.
- Murányi M, Cinar R, Kékesi O, Birkás E, Fábián G, et al. (2013) Ligand-specific regulation of the endogenous mu-opioid receptor by chronic treatment with mu-opioid peptide agonists. Biomed Res Int 2013: 501086.
- Verzillo V, Madia PA, Liu NJ, Chakrabarti S, Gintzler AR (2014) Mu-opioid receptor splice variants: Sex-dependent regulation by chronic morphine. J Neurochem 130: 790-796.
- Ingram SL, Vaughan CW, Bagley EE, Connor M, Christie MJ, et al. (1998) Enhanced opioid efficacy in opioid dependence is caused by an altered signal transduction pathway. J Neurosci 18: 10269-10276.
- Chakrabarti S, Rivera M, Yan SZ, Tang WJ, Gintzler AR (1998) Chronic morphine augments G(beta)(gamma)/Gs(alpha) stimulation of adenylyl cyclase: Relevance to opioid tolerance. Mol Pharmacol 54: 655-662.
- Chakrabarti S, Wang L, Tang WJ, Gintzler AR (1998) Chronic morphine augments adenylyl cyclase phosphorylation: Relevance to altered signaling during tolerance/dependence. Mol Pharmacol 54: 949-953.
- Gintzler AR, Chakrabarti S (2000) Opioid tolerance and the emergence of new opioid receptor-coupled signaling. Mol Neurobiol 21: 21-33.
- Chakrabarti SM, Oppermann M, Gintzler AR (2001) Chronic morphine induces the concomitant phosphorylation and altered association of multiple signaling proteins: A novel mechanism for modulating cell signaling. Proc Natl Acad Sci USA 98: 4209-4214.
- Chakrabarti S, Liu NJ, Gintzler AR (2003) Reciprocal modulation of phospholipase Cbeta isoforms: adaptation to chronic morphine. Proc Natl Acad Sci USA 100: 13686-13691.
- Chakrabarti S, Gintzler AR (2003) Phosphorylation of Gbeta is augmented by chronic morphine and enhances Gbetagamma stimulation of adenylyl cyclase activity. Brain Res Mol Brain Res 119: 144-151.
- Gintzler AR, Chakrabarti S (2004) Chronic morphine-induced plasticity among signalling molecules. Novartis Found Symp 261: 167-176.
- Chakrabarti S, Regec A, Gintzler AR (2005) Chronic morphine acts via a protein kinase Cγ-Gβ-adenylyl cyclase complex to augment phosphorylation of Gβ and Gβγ stimulatory adenylyl cyclase signaling. Mol Brain Res 138: 94-103.
- Chakrabarti S, Regec A, Gintzler AR (2005) Biochemical demonstration of mu-opioid receptor association with Gsalpha: Enhancement following morphine exposure. Brain Res Mol Brain Res 135: 217-224.
- Chakrabarti S, Gintzler AR (2007) Phosphorylation of Galphas influences its association with the micro-opioid receptor and is modulated by long-term morphine exposure. Mol Pharmacol 72: 753-760.
- Shy M, Chakrabarti S, Gintzler AR (2008) Plasticity of adenylyl cyclase-related signaling sequelae after long-term morphine treatment. Mol Pharmacol 73: 868-879.
- Gintzler AR, Chakrabarti S (2008) The ambiguities of opioid tolerance mechanisms: barriers to pain therapeutics or new pain therapeutic possibilities. J Pharmacol Exp Ther 325: 709-713.
- Chakrabarti S, Liu NJ, Zadina JE, Sharma T, Gintzler AR (2012) Pleiotropic opioid regulation of spinal endomorphin 2 release and its adaptations to opioid withdrawal are sexually dimorphic. J Pharmacol Exp Ther 340: 56-63.
- Zhang L, Loh HH, Law PY (2013) A novel noncanonical signaling pathway for the μ-opioid receptor. Mol Pharmacol 84: 844-853.
- Mao J, Price DD, Phillips LL, Lu J, Mayer DJ (1995) Increases in protein kinase C gamma immunoreactivity in the spinal cord of rats associated with tolerance to the analgesic effects of morphine. Brain Res 677: 257-267.
- Zeitz KP, Malmberg AB, Gilbert H, Basbaum AI (2001) Reduced development of tolerance to the analgesic effects of morphine and clonidine in PKC gamma mutant mice. Pain 94: 245-253.
- Wang L, Medina VM, Rivera M, Gintzler AR (1996) Relevance of phosphorylation state to opioid responsiveness in opiate naive and tolerant/dependent tissue. Brain Res 723: 61-69.
- Wang CG, Lu XF, Wei JQ, Liu H, Zhang HX, et al. (2011) Activation of the spinal extracellular signal-regulated kinase 5 signaling pathway contributes to morphine physical dependence in rats. Neurosci Lett 494: 38-43.
- Asensio VJ, Miralles A, García-Sevilla JA (2006) Stimulation of mitogen-activated protein kinase kinases (MEK1/2) by mu-, delta- and kappa-opioid receptor agonists in the rat brain: regulation by chronic morphine and opioid withdrawal. Eur J Pharmacol 539: 49-56.
- Ortiz J, Harris HW, Guitart X, Terwilliger RZ, Haycock JW, et al. (1995) Extracellular signal-regulated protein kinases (ERKs) and ERK kinase (MEK) in brain: Regional distribution and regulation by chronic morphine. J Neurosci 15: 1285-1297.
- Liang D, Li X, Clark JD (2004) Increased expression of Ca2+/calmodulin-dependent protein kinase II alpha during chronic morphine exposure. Neuroscience 123: 769-775.
- Honda M, Yoshimura N, Hikita K, Hinata N, Muraoka K, et al. (2013) Supraspinal and spinal effects of L-trans-PDC, an inhibitor of glutamate transporter, on the micturition reflex in rats. Neurourol Urodyn 32: 1026-1030.
- Storman EM, Liu NJ, Wessendorf MW, Gintzler AR (2018) Physical Linkage of Estrogen Receptor α and Aromatase in Rat: Oligocrine and Endocrine Actions of CNS-Produced Estrogens. Endocrinology 159: 2683-2697.
- Liu NJ, Murugaiyan V, Storman EM, Schnell SA, Wessendorf MW, et al. (2017) Estrogens synthesized and acting within a spinal oligomer suppress spinal endomorphin 2 antinociception: ebb and flow over the rat reproductive cycle. Pain 158: 1903-1914.
- Liu P, Rudick M, Anderson RG (2002) Multiple functions of caveolin-1. J Biol Chem 277: 41295-41298.
- Pike LJ (2009) The challenge of lipid rafts. J Lipid Res 50: 323-328.
- Simons K, Toomre D (2000) Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 1: 31-39.
- Smart EJ, Graf GA, McNiven MA, Sessa WC, Engelman JA, et al. (1999) Caveolins, liquid-ordered domains, and signal transduction. Mol Cell Biol 19: 7289-7304.
- Razani B, Rubin CS, Lisanti MP (1999) Regulation of cAMP-mediated signal transduction via interaction of caveolins with the catalytic subunit of protein kinase A. J Biol Chem 274: 26353-26360.
- Schubert W, Frank PG, Woodman SE, Hyogo H, Cohen DE, et al. (2002) Microvascular hyperpermeability in caveolin-1 (-/-) knock-out mice. Treatment with a specific nitric-oxide synthase inhibitor, L-NAME, restores normal microvascular permeability in Cav-1 null mice. J Biol Chem 277: 40091-40098.
- Woodman SE, Park DS, Cohen AW, Cheung MW, Chandra M, et al. (2002) Caveolin-3 knock-out mice develop a progressive cardiomyopathy and show hyperactivation of the p42/44 MAPK cascade. J Biol Chem 277: 38988-38997.
- Carman CV, Lisanti MP, Benovic JL (1999) Regulation of G protein-coupled receptor kinases by caveolin. J Biol Chem 274: 8858-8864.
- Lajoie P, Partridge EA, Guay G, Goetz JG, Pawling J, (2007) Plasma membrane domain organization regulates EGFR signaling in tumor cells. J Cell Biol 179: 341-356.
- Zheng YZ, Boscher C, Inder KL, Fairbank M, Loo D, et al. (2011) Differential impact of caveolae and caveolin-1 scaffolds on the membrane raft proteome. Mol Cell Proteomics 1: M110.007146.
- Lang DM, Lommel S, Jung M, Ankerhold R, Petrausch B, et al. (1998) Identification of reggie-1 and reggie-2 as plasmamembrane-associated proteins which cocluster with activated GPI-anchored cell adhesion molecules in non-caveolar micropatches in neurons. J Neurobiol 37: 502-523.
- Galbiati F, Volonte D, Gil O, Zanazzi G, Salzer JL, et al. (1998) Expression of caveolin-1 and -2 in differentiating PC12 cells and dorsal root ganglion neurons: Caveolin-2 is up-regulated in response to cell injury. Proc Natl Acad Sci USA 95: 10257-10262.
- Gaudreault SB, Blain JF, Gratton JP, Poirier J (2005) A role for caveolin-1 in post-injury reactive neuronal plasticity. J Neurochem 92: 831-839.
- Masserini M, Palestini P, Pitto M (1999) Glycolipid-enriched caveolae and caveolae-like domains in the nervous system. J Neurochem 73: 1-11.
- Cameron PL, Ruffin JW, Bollag R, Rasmussen H, Cameron RS (1997) Identification of caveolin and caveolin-related proteins in the brain. J Neurosci 17: 9520-9535.
- Ikezu T, Ueda H, Trapp BD, Nishiyama K, Sha JF, et al. (1998) Affinity-purification and characterization of caveolins from the brain: Differential expression of caveolin-1, -2, and -3 in brain endothelial and astroglial cell types. Brain Res 804: 177-192.
- Park WY, Park JS, Cho KA, Kim DI, Ko YG, et al. (2000) Up-regulation of caveolin attenuates epidermal growth factor signaling in senescent cells. J Biol Chem 275: 20847-20852.
- Park JH, Han HJ (2009) Caveolin-1 plays important role in EGF-induced migration and proliferation of mouse embryonic stem cells: involvement of PI3K/Akt and ERK. Am J Physiol Cell Physiol 297: 935-944.
- Yamamoto M, Toya Y, Schwencke C, Lisanti MP, Myers MG Jr, et al. (1998) Caveolin is an activator of insulin receptor signaling. J Biol Chem 273: 26962-26968.
- Kramer HK, Simon EJ (2000) mu and delta-opioid receptor agonists induce mitogen-activated protein kinase (MAPK) activation in the absence of receptor internalization. Neuropharmacology 39: 1707-1719.
- Li S, Seitz R, Lisanti MP (1996) Phosphorylation of caveolin by src tyrosine kinases. The alpha-isoform of caveolin is selectively phosphorylated by v-Src in vivo. J Biol Chem 271: 3863-3868.
- Aoki T, Nomura R, Fujimoto T (1999) Tyrosine phosphorylation of caveolin-1 in the endothelium. Exp Cell Res 253: 629-636.
- Lee H, Volonte D, Galbiati F, Iyengar P, Lublin DM, et al. (2000) Constitutive and growth factor-regulated phosphorylation of caveolin-1 occurs at the same site (Tyr-14) in vivo: identification of a c-Src/Cav-1/Grb7 signaling cassette. Mol Endocrinol 14: 1750-1775.
- Faulstich M, Böttcher JP, Meyer TF, Fraunholz M, Rudel T (2013) Pilus phase variation switches gonococcal adherence to invasion by caveolin-1-dependent host cell signaling. PLoS Pathog 9: 1003373.
- Jiao H, Zhang Y, Yan Z, Wang ZG, Liu G, et al. (2013) Caveolin-1 Tyr14 phosphorylation induces interaction with TLR4 in endothelial cells and mediates MyD88-dependent signaling and sepsis-induced lung inflammation. J Immunol 191: 6191-6199.
- Chen Z, Bakhshi FR, Shajahan AN, Sharma T, Mao M, et al. (2012) Nitric oxide-dependent Src activation and resultant caveolin-1 phosphorylation promote eNOS/caveolin-1 binding and eNOS inhibition. Mol Biol Cell 23: 1388-1398.
- Cao H, Courchesne WE, Mastick CC (2002) A phosphotyrosine-dependent protein interaction screen reveals a role for phosphorylation of caveolin-1 on tyrosine 14: Recruitment of C-terminal Src kinase. J Biol Chem 277: 8771-8774.
- Gintzler AR, Chakrabarti S (2006) Post-opioid receptor adaptations to chronic morphine; altered functionality and associations of signaling molecules. Life Sci 79: 717-722.
- Zhang L, Zhao H, Qiu Y, Loh HH, Law PY (2009) Src phosphorylation of micro-receptor is responsible for the receptor switching from an inhibitory to a stimulatory signal. J Biol Chem 284: 1990-2000.
- Liu W, Jiang P, Qiu L (2022) Blocking of Caveolin-1 Attenuates Morphine-Induced Inflammation, Hyperalgesia, and Analgesic Tolerance via Inhibiting NLRP3 Inflammasome and ERK/c-JUN Pathway. J Mol Neurosci 72: 1047-1057.
- Chakrabarti S, Chang A, Liu NJ, Gintzler AR (2016) Chronic opioid treatment augments caveolin-1 scaffolding: Relevance to stimulatory μ-opioid receptor adenylyl cyclase signaling. J Neurochem 139: 737-747.
- Ringkamp M, Eschenfelder S, Grethel EJ, Häbler HJ, Meyer RA, et al. (1999) Lumbar sympathectomy failed to reverse mechanical allodynia- and hyperalgesia-like behavior in rats with L5 spinal nerve injury. Pain 79: 143-153.
- Li Y, Dorsi MJ, Meyer RA, Belzberg AJ (2000) Mechanical hyperalgesia after an L5 spinal nerve lesion in the rat is not dependent on input from injured nerve fibers. Pain 85: 493-502.
- Leong ML, Gu M, Speltz-Paiz R, Stahura EI, Mottey N, et al. (2011) Neuronal loss in the rostral ventromedial medulla in a rat model of neuropathic pain. J Neurosci 31: 17028-17039.
- Ho Kim S, Mo Chung J (1992) An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50: 355-363.
- Kim KJ, Yoon YW, Chung JM (1997) Comparison of three rodent neuropathic pain models. Exp Brain Res 113: 200-206.
- Blenk KH, Häbler HJ, Jänig W (1997) Neomycin and gadolinium applied to an L5 spinal nerve lesion prevent mechanical allodynia-like behaviour in rats. Pain 70: 155-165.
- Gintzler AR, Liu NJ (2019) Harnessing endogenous opioids for pain relief: Fantasy vs reality. J Opioid Manag 16: 67-72.
- Pelissier T, Paeile C, Soto-Moyano R, Saavedra H, Hernández A (1990) Analgesia produced by intrathecal administration of the kappa opioid agonist, U-50,488H, on formalin-evoked cutaneous pain in the rat. Eur J Pharmacol 190: 287-293.
- Sullivan AF, Dickenson AH (1991) Electrophysiologic studies on the spinal antinociceptive action of kappa opioid agonists in the adult and 21-day-old rat. J Pharmacol Exp Ther 256: 1119-1125.
- Dawson-Basoa ME, Gintzler AR (1996) Estrogen and Progesterone activate spinal kappa-opiate receptor analgesic mechanisms. Pain 64: 607-615.
- Powell KJ, Ma W, Sutak M, Doods H, Quirion R, et al. (2000) Blockade and reversal of spinal morphine tolerance by peptide and non-peptide calcitonin gene-related peptide receptor antagonists. Br J Pharmacol 131: 875-884.
- Liu NJ, Chakrabarti S, Gintzler AR (2004) Chronic morphine-induced loss of the facilitative interaction between vasoactive intestinal polypeptide and delta-opioid: Involvement of protein kinase C and phospholipase Cbetas. Brain Res 1010: 1-9.
- Bhargava HN, Villar VM (1991) Tolerance-dependence and serum elimination of morphine in rats implanted with morphine pellets. Gen Pharmacol 22: 1033-1042.
- Liu NJ, Storman EM, Gintzler AR (2019) Pharmacological Modulation of Endogenous Opioid Activity to Attenuate Neuropathic Pain in Rats. J Pain 20: 235-243.
- Liu NJ, Gintzler AR (2013) Spinal Endomorphin 2 Antinociception and the Mechanisms That Produce It Are Both Sex- and Stage of Estrus Cycle-Dependent in Rats. J Pain 14: 1522-1530.
- Liu NJ, Murugaiyan V, Storman EM, Schnell SA, Kumar A, et al. (2017) Plasticity of Signaling by Spinal Estrogen Receptor α, κ-Opioid Receptor, and Metabotropic Glutamate Receptors over the Rat Reproductive Cycle Regulates Spinal Endomorphin 2 Antinociception: Relevance of Endogenous-Biased Agonism. J Neurosci 37: 11181-11191.
- Piñeyro G, Archer-Lahlou E (2007) Ligand-specific receptor states: Implications for opiate receptor signalling and regulation. Cell Signal 19: 8-19.
- Law PY, Hom DS, Loh HH (1982) Loss of opiate receptor activity in neuroblastoma X glioma NG108-15 hybrid cells after chronic opiate treatment. A multiple-step process. Mol Pharmacol 22: 1-4.
- Sharma SK, Klee WA, Nirenberg M (1977) Opiate-dependent modulation of adenylate cyclase. Proc Natl Acad Sci USA 74: 3365-3369.
- Srere PA, Sumegi B (1994) Processivity and fatty acid oxidation. Biochem Soc Trans 22: 446-450.
- Cheung CW, Cohen NS, Raijman L (1989) Channeling of urea cycle intermediates in situ in permeabilized hepatocytes. J Biol Chem 264: 4038-4044.
- Campanella ME, Chu H, Low PS (2005) Assembly and regulation of a glycolytic enzyme complex on the human erythrocyte membrane. Proc Natl Acad Sci USA 102: 2402-2407.
- Robinson JB Jr, Srere PA(1985) Organization of Krebs tricarboxylic acid cycle enzymes in mitochondria. J Biol Chem 260: 10800-10805.
- Wellen KE, Lu C, Mancuso A, Lemons JM, Ryczko M, et al. (2010) The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev 24: 2784-2799.
- Bannister K, Dickenson AH (2016) What the brain tells the spinal cord. Pain 157: 2148-2151.
- Bannister K, Dickenson AH (2016) What do monoamines do in pain modulation? Curr Opin Support Palliat Care 10: 143-148.
- Devulder J (1997) Hyperalgesia induced by high-dose intrathecal sufentanil in neuropathic pain. J Neurosurg Anesthesiol 9): 146-148.
- de Conno F, Caraceni A, Martini C, Spoldi E, Salvetti M, et al. (1991) Hyperalgesia and myoclonus with intrathecal infusion of high-dose morphine. Pain 47: 337-339.
- Ali NM (1986) Hyperalgesic response in a patient receiving high concentrations of spinal morphine. Anesthesiology 65: 449.
- Arnér S, Rawal N, Gustafsson L (1988) Clinical experience of long-term treatment with epidural and intrathecal opioids--a nationwide survey. Acta Anaesthesiol Scand 32: 253-259.
- Mayer DJ, Mao J, Price DD (1995) The association of neuropathic pain, morphine tolerance and dependence, and the translocation of protein kinase C. NIDA Res Monogr 147: 269-298.
- Vanderah TW, Suenaga NM, Ossipov MH, Malan TP Jr, Lai J, et al. (2001) Tonic descending facilitation from the rostral ventromedial medulla mediates opioid-induced abnormal pain and antinociceptive tolerance. J Neurosci 21: 279-286.
- Audet N, Paquin-Gobeil M, Landry-Paquet O, Schiller PW, Piñeyro G (2005) Internalization and Src activity regulate the time course of ERK activation by delta opioid receptor ligands. J Biol Chem 280: 7808-7816.
Citation: Liu N-J, Li H (2022) Disrupting Spinal Caveolin-1 Scaffolding Prevents as well as Reverses Tolerance to Opioid Antiallodynia in Treating Neuropathic Pain in Rats. J Reprod Med Gynecol Obstet 7: 0103.
Copyright: © 2022 Nai-Jiang Liu, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.