Journal of Alcoholism Drug Abuse & Substance Dependence Category: Medical
Type: Research Article
Does Vitamin E Protect Against Hepatic Oxidative Stress During Alcohol Metabolism in Rodent Liver Cell Lines? - An EPR Study
*Corresponding Author(s):
Barry J PotterDepartment Of Physiology, School Of Medicine, Louisiana Health, 1901 Perdido Street, New Orleans, LA 70112-1393, United States
Tel:+1 5045683385,
Email:bpotte@lsuhsc.edu
Received Date: Jul 23, 2015
Accepted Date: Sep 07, 2015
Published Date: Sep 30, 2015
Abstract
Alcohol intoxication is known to be associated with free radical generation and cellular oxidative stress. In consequence, treatment of recovering chronic alcoholic subjects frequently includes administration of a variety of antioxidant substances, such as vitamin E (alpha-tocopherol). However, recent controversial studies have suggested that vitamin E supplementation may not be cardioprotective, but no such data are available for hepatic consequences. The aim of this study was therefore to examine the protective effects of vitamin E in an in vitro Electron Paramagnetic Resonance (EPR) study of alcohol-mediated free radical generation. Two cell lines were used, rat FtO2B cells and mouse RAW 264.7, to represent the major cell populations of the liver - parenchymal (hepatocytes) and non-parenchymal (Kupffer cells) -cells respectively. Both cell lines express all of the major functions of normal liver cells & are therefore considered to be representative of a typical liver. Cells were incubated in medium containing 100mM ethanol and the presence or absence of a range of concentrations of either vitamin E, or the mimetic Trolox, and the free radical trapped using the spin trapping agent CPH. The radical adducts were then quantitated at room temperature by double integration of the X-band spectra obtained from a Bruker EMX spectrometer. All doses of vitamin E reduced free radical generation by at least 35-45% in both cell types in both acute and chronic studies. By contrast, however, the water-soluble mimetic Trolox acutely increased free radical production in Ft02B cells and had no protective effect after 1 hour pretreatment. In RAW 264.7 cells, it had no acute protective effects, but increased free radical production following 1 hour pretreatment. These results suggest that the possible hepatoprotective effect of vitamin E is related to its lipid solubility and that its presence in the hepatic cytosol may stimulate free radical formation, leading to a nullification of the protective effect.
INTRODUCTION
Alcohol is rapidly metabolized to acetaldehyde by alcohol dehydrogenase and then to acetic acid and acetyl-CoA (which can enter the citric acid cycle). This metabolism is associated with a marked change in the redox potential and transformation of xanthine dehydrogenase to xanthine oxidase, resulting in free radical production [1]. Continuous alcohol intake will also result in induction of cytochrome P450 2E1 and consequential lipid peroxidation and generation of Reactive Oxygen Species (ROS) causing damage to cellular components [2]. Chronic alcohol abuse is known to result in damage to multiple organs, especially the liver and it has long been accepted that a factor in the pathogenesis of alcoholic liver disease is oxidative stress resulting from the production of free radical species. As it is generally accepted that this damage is continuous during drinking but at a low level, we have previously examined the effect of use of an organic peroxide, tertiary-Butyl Hydroperoxide (tBH) to investigate increased sensitivity to ethanol in rodent hepatocytes and cell lines [3]. Some previous studies clearly demonstrated that alcohol related damage was associated with lipid peroxidation and that free iron is possibly involved in this pathway [4].
While the liver cells contain anti-oxidant molecules such as glutathione, vitamins C and E, and antioxidant enzymes, these become depleted with continuous alcohol intake [5,6]. The consequential change in redox imbalance caused by ROS generation has been observed in patients with alcoholic liver disease [7] but attempts to ameliorate these disturbances through the administration of antioxidants has met with mixed results [1,8,9]. In contrast to this, many animal model studies (principally with rodents) have shown that these antioxidants and vitamin E in particular do have mitigating effects on free radical damage of the liver related to alcohol abuse [10-14]. Much of the damage appears to be associated with peroxidation of lipid membranes as use of N,N-Diphenyl-P-Phenlylenediamine (DPPD) a lipid radical scavenger has been shown to prevent oxidation and liver cell death in α-tocopherol (a form of vitamin E) deficient rats and cultured hepatocytes [3,15].
Antioxidant supplementation for a wide variety of diseases states has been widely practiced. However, this continues to this day to be controversial. Vitamin E has been extensively studied as a cardio-protectant for many years, but there is little agreement as to its effectiveness [9,16-19]. In general, plasma antioxidants are low in patients with alcoholic liver disease [8] and with non-alcoholic fatty liver [20]. While clearly an antioxidant, vitamin E has shown mixed results in clinical trials on a variety of liver disease patients [21-25]. Experimental models tend to show a clearer picture, with vitamin E reducing lipid peroxidation [26,27] but have usually either studied acute or overnight effects and have not dissected the liver cell populations to determine whether there is a differential effect. It was the aim of this study, therefore to examine the effects of a stressor (tBH) on cells representing hepatocytes and non-parenchymal cells (Kupffer cells) incubated in alcohol containing medium and to assess both the short-term (4 hours) to long-term (7 days) protective effect of a range of concentrations of vitamin E.
While the liver cells contain anti-oxidant molecules such as glutathione, vitamins C and E, and antioxidant enzymes, these become depleted with continuous alcohol intake [5,6]. The consequential change in redox imbalance caused by ROS generation has been observed in patients with alcoholic liver disease [7] but attempts to ameliorate these disturbances through the administration of antioxidants has met with mixed results [1,8,9]. In contrast to this, many animal model studies (principally with rodents) have shown that these antioxidants and vitamin E in particular do have mitigating effects on free radical damage of the liver related to alcohol abuse [10-14]. Much of the damage appears to be associated with peroxidation of lipid membranes as use of N,N-Diphenyl-P-Phenlylenediamine (DPPD) a lipid radical scavenger has been shown to prevent oxidation and liver cell death in α-tocopherol (a form of vitamin E) deficient rats and cultured hepatocytes [3,15].
Antioxidant supplementation for a wide variety of diseases states has been widely practiced. However, this continues to this day to be controversial. Vitamin E has been extensively studied as a cardio-protectant for many years, but there is little agreement as to its effectiveness [9,16-19]. In general, plasma antioxidants are low in patients with alcoholic liver disease [8] and with non-alcoholic fatty liver [20]. While clearly an antioxidant, vitamin E has shown mixed results in clinical trials on a variety of liver disease patients [21-25]. Experimental models tend to show a clearer picture, with vitamin E reducing lipid peroxidation [26,27] but have usually either studied acute or overnight effects and have not dissected the liver cell populations to determine whether there is a differential effect. It was the aim of this study, therefore to examine the effects of a stressor (tBH) on cells representing hepatocytes and non-parenchymal cells (Kupffer cells) incubated in alcohol containing medium and to assess both the short-term (4 hours) to long-term (7 days) protective effect of a range of concentrations of vitamin E.
MATERIALS AND METHODS
Cell culture
Two cell lines were used, FtO2B (a rat hepatoma cell line) and RAW 264.7, to represent parenchymal and non-parenchymal liver cells, which comprise more than 75% of the total cell population of a normal liver [approximately 60% are hepatocytes and 10-15% hepatic macrophages/Kupffer cells]. FtO2B cells which are a rat hepatoma cell line were a kind gift from Dr. J Lancaster Jr. RAW 264.7 cells were purchased from ATCC. This cell line is a mouse peritoneal macrophage cell line established from a tumor induced by Abelson murine leukemia virus. The rat hepatoma cell line FtO2B was grown at 37°C in a 5% CO2atmosphere in Dulbecco’s modified Eagle’s medium/Ham’s F12 medium (1:1) supplemented with 10% non-heat-inactivated bovine calf serum, 2mM glutamine, 50 U/ml penicillin-streptomycin, and 0.2% sodium bicarbonate. FTO2B cells (2 × 105/ml) were plated in 12-well plates and allowed to adhere overnight before an experiment. Cells were used for experiments only when they were 60% confluent or more and did not reach more than 90% confluence over 7 days experimentation. The RAW 264.7 cells, representing non-parenchymal macrophages, were cultured in RPMI 1640 medium similarly supplemented with 10% non-heat-inactivated bovine calf serum, 2mM glutamine, 50 U/ml penicillin-streptomycin, and 0.2% sodium bicarbonate (Sigma-Aldrich). RAW 264.7 cells (2 × 105/ml) were again plated in 12-well plates and allowed to adhere overnight before experimentation.
Alcohol metabolism
To assess the capability of these cells to metabolize ethanol, cells were incubated in the presence of 100mM ethanol and the rate of disappearance from the culture medium quantitated using an alcohol determination kit (Sigma-Aldrich). Evaporation losses were determined using 12-well plates containing supplemented medium and alcohol alone. Ethanol-containing medium was changed every 24 hours and the rate estimated daily for 1 week.
Free radical generation
Preliminary data on free radical generation was generated through assessment of Thiobarbituric Acid Reactive Substances (TBARS). While alcohol is considered to be a stressor, it is generally considered that severe oxidative stress requires a second insult (the “second hit” theory) [28]. FtO2B cells were incubated for 1 hour at 37oC in the presence of 100mM ethanol, 100 µM ferrous sulfate (Fe), 100 µM Fe + 100mM ethanol, 400 µM t-Butyl Hydroperoxide (tBH - Sigma-Aldrich) ± 100mM ethanol. Of these treatments, tBH was selected for the oxidative stress experiments because of a well-defined free radical and cell killing response that we had previously determined for rat hepatocytes & FtO2B cells [3]. Viability of FtO2B cells at various concentrations of 100mM EtOH, or medium alone were assessed after 24 hour culture using an assay for lactate dehydrogenase in the culture supernatant and lysed cells.
Experiments to determine the efficacy of vitamin E (α-tocopherol) as an antioxidant were then performed on these cell cultures at time points ranging from acute (2 hours ) incubations up to 7 days in cell culture. The optimal range of concentrations of vitamin E was determined from an initial acute study using FtO2B cells and a 2 hour incubation (data not shown). 12-well plates were then incubated for either 4 hours, 16 hours (overnight) or 7 days in the presence or absence of ethanol and/or a range of vitamin E concentrations (0 - 100 µM). 10 wells of each plate were used for free radical determinations and 2 for viability assays. Experiments were also carried out using the same protocol but adding a range of Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid - Hoffman-LaRoche) concentrations (0 -250 µM) , a more hydrophilic analog of vitamin E, instead of vitamin E. Medium in all culture wells was changed daily.
Experiments to determine the efficacy of vitamin E (α-tocopherol) as an antioxidant were then performed on these cell cultures at time points ranging from acute (2 hours ) incubations up to 7 days in cell culture. The optimal range of concentrations of vitamin E was determined from an initial acute study using FtO2B cells and a 2 hour incubation (data not shown). 12-well plates were then incubated for either 4 hours, 16 hours (overnight) or 7 days in the presence or absence of ethanol and/or a range of vitamin E concentrations (0 - 100 µM). 10 wells of each plate were used for free radical determinations and 2 for viability assays. Experiments were also carried out using the same protocol but adding a range of Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid - Hoffman-LaRoche) concentrations (0 -250 µM) , a more hydrophilic analog of vitamin E, instead of vitamin E. Medium in all culture wells was changed daily.
Free radical determinations
Free radical determinations were carried out on suspensions of cells in medium, incubated with 2.0mM α-(4-Pyridyl 1-oxide)-N-tert-butylnitrone (POBN - Enzo Life Sciences), a cell permeable hydrophilic spin trapping agent. This agent traps both superoxide and hydroxyl free radicals [29] but superoxide adducts tend to be unstable, forming false hydroxyl complexes. However the current study was to determine total free radical generation in response to ethanol and other stressors.
The standard procedure for ROS determination was to incubate the wells containing FtO2B or RAW 264.7 cells with tBH and POBN for 30 minutes at 37°C and then remove the entire contents of the wells and snap-freeze them in Liquid Nitrogen (LN2). Samples were then stored in LN2 until EPR spectroscopy was performed. For EPR measurements, snap-frozen samples were rapidly thawed at 37°C and aspirated into glass capillaries (ID 1mm) and spectral intensity read at room temperature using a Bruker EMX spectrometer & quantitation of this by double integration of the spectra. The EPR spectrum settings were as follows: modulation amplitude 1.0 gauss, scan time 83 seconds, time constant 163 msec and microwave power 40 mW, field sweep 60 gauss, microwave frequency 9.78 GHz (X-band), receiver gain 5 x 103, center field 3485 gauss, as described previously [30]. ROS quantification from the EPR spectra was determined by double integration of the peaks, Day-to-day reproducibility was maintained through use of a 100 µM solution of TEMPOL (4-Hydroxy-2,2,6,6-tetramethylpiperidinyloxy - Enzo Life Sciences), a natural spin adduct. All results were normalized to the sample protein concentration. Values are expressed as arbitrary values or % control values.
The standard procedure for ROS determination was to incubate the wells containing FtO2B or RAW 264.7 cells with tBH and POBN for 30 minutes at 37°C and then remove the entire contents of the wells and snap-freeze them in Liquid Nitrogen (LN2). Samples were then stored in LN2 until EPR spectroscopy was performed. For EPR measurements, snap-frozen samples were rapidly thawed at 37°C and aspirated into glass capillaries (ID 1mm) and spectral intensity read at room temperature using a Bruker EMX spectrometer & quantitation of this by double integration of the spectra. The EPR spectrum settings were as follows: modulation amplitude 1.0 gauss, scan time 83 seconds, time constant 163 msec and microwave power 40 mW, field sweep 60 gauss, microwave frequency 9.78 GHz (X-band), receiver gain 5 x 103, center field 3485 gauss, as described previously [30]. ROS quantification from the EPR spectra was determined by double integration of the peaks, Day-to-day reproducibility was maintained through use of a 100 µM solution of TEMPOL (4-Hydroxy-2,2,6,6-tetramethylpiperidinyloxy - Enzo Life Sciences), a natural spin adduct. All results were normalized to the sample protein concentration. Values are expressed as arbitrary values or % control values.
Statistics
Each type of cell was analyzed separately and are only shown together to highlight changes in response to free radical generation caused by antioxidant treatment. Each of the 10 wells on every plate was analyzed separately for free radical generation at each time point (10 individual readings per time point). Statistical analysis was performed using ANOVA followed by t-tests with the Bonferroni inequality was used to determine significance. A minimum probability value of p<0.05 was used to determine statistical significance.
RESULTS
Ethanol metabolism
Ethanol metabolism by FtO2B cells in culture was steady and sustained. As can be seen in (Figure 1A), alcohol metabolism was approximately linear over 5 hours; the corrected catabolic rate being estimated at 9.50 + 0.57 µmoles/mg cell protein. As anticipated, ethanol disappearance from the tissue culture wells accounted for approximately 25%/day; the total change in alcohol content/hour being shown in (Figure 1B). No significant metabolism of ethanol by RAW 264.7 cells was observed during this period. Although there was a trend towards minimal metabolism, values for ethanol disappearance were not significantly greater than the values observed for total ethanol loss from the flasks.
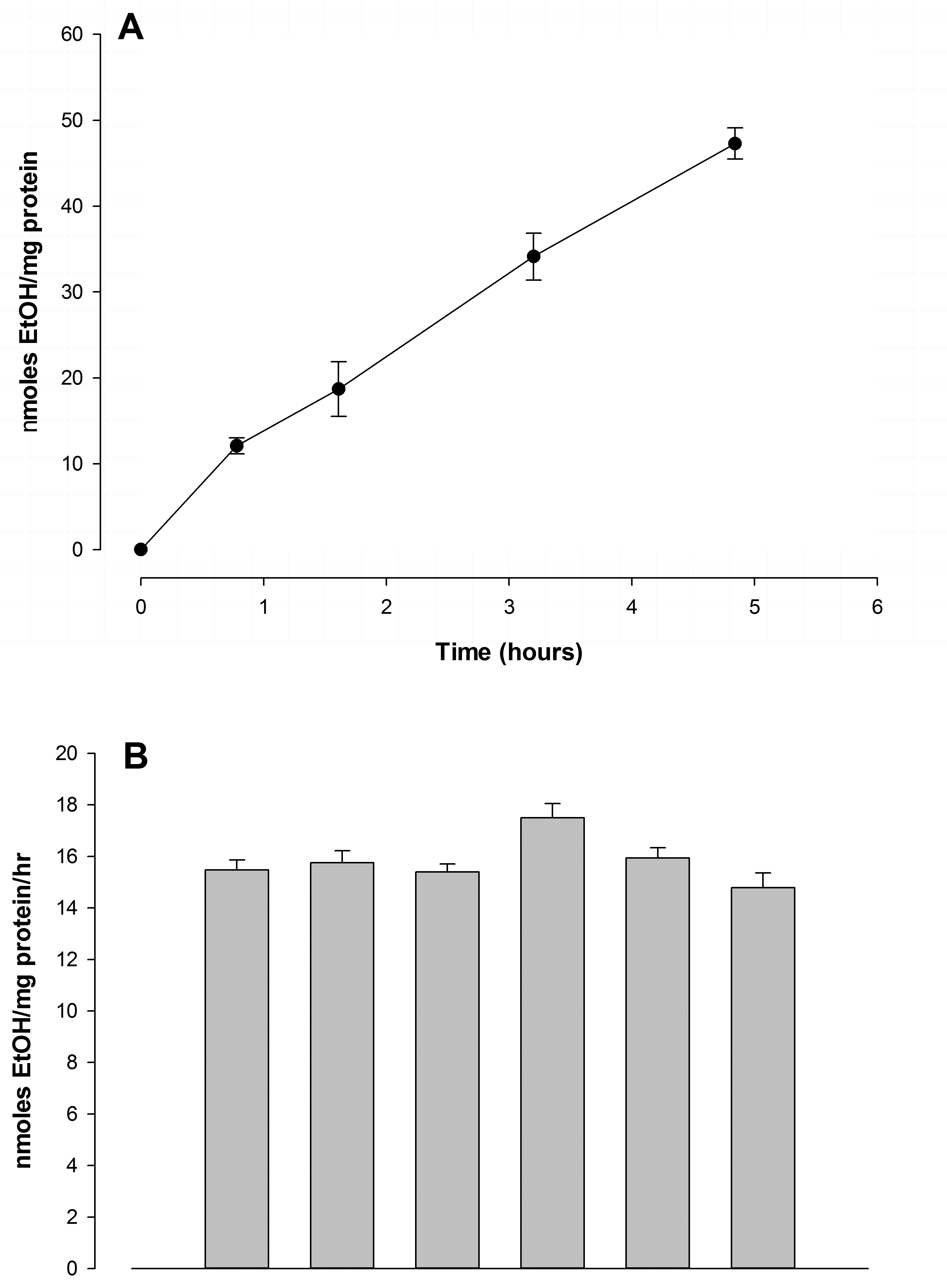
Figure 1: Ethanol metabolism by FtO2B cells in culture.
A.12-well plates containing FtO2B cells grown to 90% confluence were incubated at 37°C in a 5% CO2atmosphere in 100mM ethanol in Dulbecco’s modified Eagle’s medium/Ham’s F12 medium (1:1) supplemented with 10% non-heat-inactivated bovine calf serum, 2mM glutamine, 50 U/ml penicillin-streptomycin, and 0.2% sodium bicarbonate. Aliquots of the supernatant were aspirated and used to determine residual alcohol content. These were then corrected for evaporation using values obtained from 12-well plates containing the alcohol solution. Values represent the means+ SEM of at least 12 determinations.
B. 12 well plates containing FtO2B cells grown to 60% confluence were incubated at 37°C in a 5% CO2atmosphere in medium containing 100mM ethanol for 1-7 days. The alcohol containing medium was changed daily. Aliquots were aspirated and ethanol concentrations assayed using a commercial kit (Sigma-Aldrich). Loss through ethanol evaporation was determined using plates containing ethanol and medium alone. Values represent the mean + SEM of 10 determinations. The other 2 wells were chosen randomly and viability of the cells assessed by trypan blue exclusion. Cells were then scraped, washed twice with phosphate-buffered saline and total protein per well quantitated.
The presence of 100mM ethanol, while clearly being metabolized by FtO2B cells, did not generate significant quantities of free radicals when compared to cells incubated in medium alone, as may be seen for the control values in (Figure 2). Viability of these cells as determined by trypan blue exclusion & medium LDH values gave slightly different values. Trypan blue dye exclusion consistently gave 96% ± 2% for all cultures in the presence or absence of alcohol over 7 days culture. However, LDH values in cells incubated in the presence of ethanol suggested a reduction in viability from 98% after 4 hours to 93 + 3% after 7 days incubation. Interpretation of these values is difficult since the cells grew to almost 90% confluence during this time period. This change in viability was not statistically different from that seen with cells cultured in medium alone. Addition of 400 µM t-Butyl Hydroperoxide (tBH) to these cells resulted in a 2500 times increase in free radicals irrespective of the presence of ethanol (p = NS). Treatment with 5 µM cyanamide, an inhibitor of acetaldehyde metabolism [31], had no effect on free radical production. Addition of 100 µM 4-Methylpyrazole (4-MP [32]) significantly decreased free radical generation, but only in cells cultured in ethanol-containing medium.B. 12 well plates containing FtO2B cells grown to 60% confluence were incubated at 37°C in a 5% CO2atmosphere in medium containing 100mM ethanol for 1-7 days. The alcohol containing medium was changed daily. Aliquots were aspirated and ethanol concentrations assayed using a commercial kit (Sigma-Aldrich). Loss through ethanol evaporation was determined using plates containing ethanol and medium alone. Values represent the mean + SEM of 10 determinations. The other 2 wells were chosen randomly and viability of the cells assessed by trypan blue exclusion. Cells were then scraped, washed twice with phosphate-buffered saline and total protein per well quantitated.
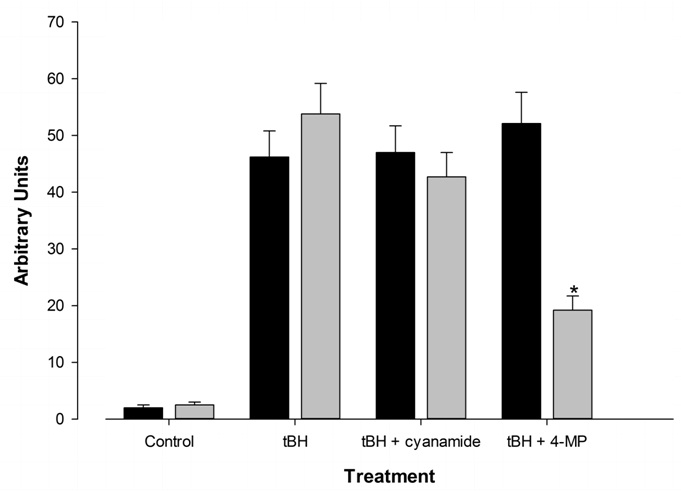
Figure 2: Effect of ethanol metabolic inhibitors on free radical generation by FtO2B cells in culture following oxidative stress from 400 µM t-butyl hydroperoxide (tBH).
12 well plates containing FtO2B cells in cultured in the presence or absence of 100mM ethanol were incubated for 30 minutes in the presence of 400 µM t-Butyl Hydroperoxide (tBH) + the alcohol metabolic inhibitors cyanamide (5 µM) and 4-methyl pyrazole (100 µM), and 2 mM POBN. The supernatants were then aspirated and snap-frozen in Liquid Nitrogen (LN2) until free radical determination in a Bruker EMX EPR spectrometer at 9.78 GHz. Alcohol-containing medium, medium alone. Values represent the mean + SEM of 10 determinations. *p< 0.001 vs. medium alone containing tBH.
Antioxidant effects of vitamin E
Free radical production, elicited by the addition of tBH remained maximal throughout 7 days in culture. The addition of vitamin E to FtO2B cells cultured in the presence of alcohol from 4 hours to 7 days significantly reduced free radical production caused by the addition of t-BOOH. As may be seen in (Figure 3), maximal free radical inhibition occurred throughout the range of vitamin E doses administered following overnight incubation. This antioxidant effect was significantly less pronounced when similar treatment was applied to RAW 264.7 cells, only reaching significant values at the higher concentrations following 4 hours incubation (Figure 3A). Overnight culture of both cell types resulted in significant lowering of free radical production (Figure 3B); reducing generation in FtO2B cells to almost normal levels. Vitamin E was less effective in RAW 264.7 cells lowering free radical generation by approximately 40%. Following 7 days culture, vitamin E tended to have less of an antioxidant effect for FtO2B cells (Figure 3C). Never the less, vitamin E still appeared to have a protective effect against free radical production.
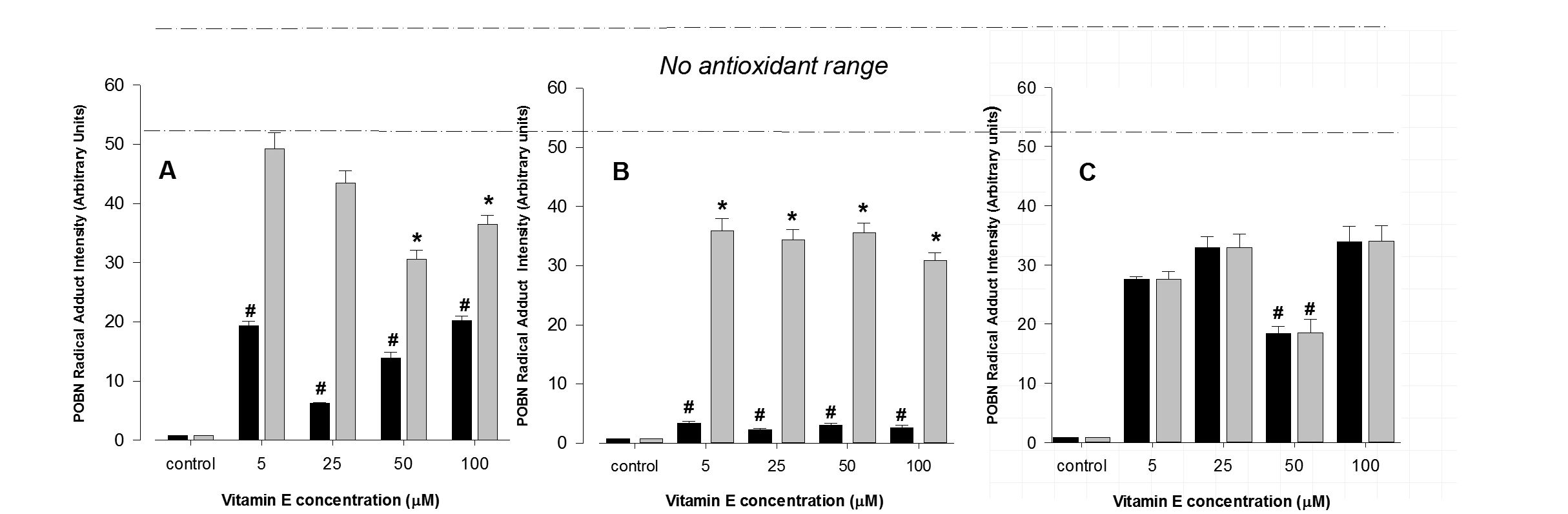
Figure 3: Acute antioxidant effect of vitamin E on FtO2B and RAW 264.7 cells during oxidative stress generation.
12 well plates containing FtO2B cells or RAW 264.7 cells in culture in the presence of 100mM ethanol and 400 µM t-Butyl Hydroperoxide (tBH) were incubated for 4 hours (A), 16 hours (B) or 7 days (C) over a range of concentrations of vitamin E (0 - 100 µM). Cells were then incubated for 30 minutes in the presence of 400 µM t-Butyl Hydroperoxide (tBH) and 2mM POBN. The supernatants were then aspirated and snap-frozen in Liquid Nitrogen (LN2) until free radical determination in a Bruker EMX EPR spectrometer at 9.78 GHz.
FtO2B cells; RAW 264.7 cells. *p
FtO2B cells; RAW 264.7 cells. *p
Antioxidant effects of a vitamin E mimetic
The effect of a commercial vitamin E mimetic (Trolox) was investigated for amelioration of free radical formation from both FtO2B and RAW 264.7 cells. Cells were incubated for 4 hours and overnight in the presence of a range of Trolox concentrations. However, unlike vitamin E, there was no significant change in free radical production at any Trolox concentration (Figure 4A) from untreated FtO2B or RAW 264.7 cells following a 4 hour incubation. Overnight cultures resulted in a modest (p < 0.05) protective effect for FtO2B cells at concentrations of 10 and 50 µM Trolox but an increased free radical formation in RAW 264.7 cells at the same concentrations (Figure 4B). No results for 7 day cultures in the presence of Trolox have been included due to non-reproducibility & low viability.
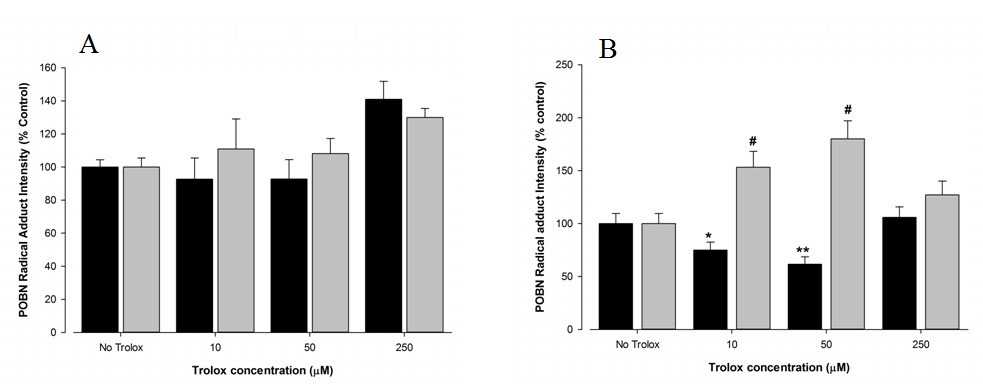
Figure 4: Acute antioxidant effect of Trolox on FtO2B and RAW 264.7 cells during oxidative stress generation.
12 well plates containing FtO2B cells or RAW 264.7 cells in culture in the presence of 100mM ethanol were incubated for 4 hours (A), or 16 hours (B), over a range of concentrations of Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid). Cells were then incubated for 30 minutes in the presence of 400 µM t-Butyl Hydroperoxide (tBH) and 2mM POBN. The supernatants were then aspirated and snap-frozen in Liquid Nitrogen (LN2) until free radical determination in a Bruker EMX EPR spectrometer at 9.78 GHz. FtO2B cells; RAW 264.7 cells. *p< 0.05 vs RAW 264.7; # p < 0.05 vs control values.
DISCUSSION
Vitamin E, discovered in 1922 [33] has long been known to be an essential nutrient and have antioxidant and pro-oxidant properties [34]. In particular it is known to prevent mitochondrial dysfunction in response to tBH induced oxidative stress and reduce membrane lipid oxidation in rodents [35]. Experimentally, ethanol intoxication has been shown to be associated with free radical generation [36]. α-tocopherol supplementation to rats chronically exposed to ethanol has been shown to prevent the formation of Mallory bodies and inflammatory infiltration of the liver but not increased apoptosis, inspite of increased antioxidant gene expression [6]. Several groups have shown that subjects with alcoholic liver disease have been shown to have significantly lower plasma vitamin E levels [9,37,38] and a higher degree of redox imbalance [7]. However, the results of long-term vitamin E supplementation have been controversial and inconsistent [5,14,39,40] although positive results have been reported in patients with non-alcoholic fatty liver [20,21].
Because of these inconsistent results, we have studied the effect of vitamin E supplementation in tissue culture using two cell lines, to investigate possible differential cell responses. This study is part of a larger investigation into the effects of free radical generation following alcohol intoxication and the transition from cell signaling at low concentrations to cellular damage at higher levels using Electron Paramagnetic Resonance (EPR) techniques. The 2 cell lines were chosen to reflex the two major cell populations in the liver; parenchymal cells (FtO2B) and non-parenchymal cells (RAW 264.7). While these are cell lines & not primary cultures of liver cell populations, both cell lines clearly demonstrate most normal liver cell functions and have been used by many investigators in hepatology research. Since the liver hepatocytes are the primary site of ethanol metabolism, we examined the rate of disappearance from the media (adjusted for normal evaporation). The FtO2B cells in this study clearly metabolized ethanol at a constant rate throughout the 7 day study. The rate of ethanol metabolism is similar to that seen in rat hepatocytes [41]. Furthermore, this rate of catabolism appeared constant over 7 days of cell culture with minimal cell death (< 10%). However, a dose of 400 µM tBH was chosen from a dose response curve for the oxidative stress because it had been previously shown to result in 40-50% loss of viability following 120 minutes incubation (Obih et al.,). tBH itself has been used in lipid per oxidation studies and is often used in antioxidant experiments because of its relatively slow generation of free radicals [42,43]. The relationship between free radical generation and alcohol metabolism becomes apparent when ethanol metabolic inhibitors are added: the addition of 4-methylpyrazole to cells cultured in ethanol significantly lowers the free radical generation but not cyanamide. As this is inhibiting alcohol dehydrogenase, but not acetaldehyde dehydrogenase, this implies that free radical production does not occur during the first step of alcohol catabolism, or oxidative stress in these cells is unaffected by ethanol breakdown.
The addition of vitamin E to FtO2B cells in culture clearly has a significant direct protective effect at all concentrations used. Surprisingly it was most effective after an overnight culture and slightly less effective at 7 days. This probably reflects a slower distribution within the cells to where it can exert its beneficial action. For RAW 264.7 cells, however, 5 µM had no protective effect when incubated for 4 hours. After that period, overnight incubation resulted in a dose-dependent tendency for protection against free radicals. After 7 days incubation, protection against free radicals was effectively the same as for the FtO2B cells. These findings correlate well with those observed by Jordao and co-workers in rats [26], but not in patients with alcoholic cirrhosis [5].
Because of the time lag for maximum protection in the FtO2B cells, trolox was substituted for α-tocopherol in one set of experiments with FtO2B and RAW 264.7 cells. Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid) is a water-soluble vitamin E mimetic that will nevertheless partition into liposomes [44] and has been used as a commercial standard for antioxidant activity. Unlike α-tocopherol, it had no protective effect at concentrations from 10-250 µM when incubated with FtO2B and RAW 264.7 cells for 4 hours in the presence of ethanol. Overnight culture resulted in some protection for FtO2B cells at concentrations of 10 and 50 µM, but pro-oxidant activity for the RAW 264.7 cells at the same concentrations. Why this occurred is not clear, but it is known that trolox can exhibit pro-oxidant activity in the presence of transition metals [45] and free iron may be released from the cells to generate a Fenton-type reaction. Cell cultures of tumorigenic cells have also shown minimal protection against ROS and impaired cell survival [46].
From these data it is clear that protection from free radicals in alcoholic liver disease is dependent not only on the antioxidant, but also the cell type. Naturally occurring vitamin E is a composite of 4 tocopherols and 4 tocotrienols, of which a-tocopherol is the most abundant. Based on our data, it is possible that one of the other 7 compounds may prove to be equally effective, if not more so. Furthermore, cell differences in handling of vitamin E may make true protection problematic unless it can be targeted. Further research is clearly needed into these findings from the above research.
Because of these inconsistent results, we have studied the effect of vitamin E supplementation in tissue culture using two cell lines, to investigate possible differential cell responses. This study is part of a larger investigation into the effects of free radical generation following alcohol intoxication and the transition from cell signaling at low concentrations to cellular damage at higher levels using Electron Paramagnetic Resonance (EPR) techniques. The 2 cell lines were chosen to reflex the two major cell populations in the liver; parenchymal cells (FtO2B) and non-parenchymal cells (RAW 264.7). While these are cell lines & not primary cultures of liver cell populations, both cell lines clearly demonstrate most normal liver cell functions and have been used by many investigators in hepatology research. Since the liver hepatocytes are the primary site of ethanol metabolism, we examined the rate of disappearance from the media (adjusted for normal evaporation). The FtO2B cells in this study clearly metabolized ethanol at a constant rate throughout the 7 day study. The rate of ethanol metabolism is similar to that seen in rat hepatocytes [41]. Furthermore, this rate of catabolism appeared constant over 7 days of cell culture with minimal cell death (< 10%). However, a dose of 400 µM tBH was chosen from a dose response curve for the oxidative stress because it had been previously shown to result in 40-50% loss of viability following 120 minutes incubation (Obih et al.,). tBH itself has been used in lipid per oxidation studies and is often used in antioxidant experiments because of its relatively slow generation of free radicals [42,43]. The relationship between free radical generation and alcohol metabolism becomes apparent when ethanol metabolic inhibitors are added: the addition of 4-methylpyrazole to cells cultured in ethanol significantly lowers the free radical generation but not cyanamide. As this is inhibiting alcohol dehydrogenase, but not acetaldehyde dehydrogenase, this implies that free radical production does not occur during the first step of alcohol catabolism, or oxidative stress in these cells is unaffected by ethanol breakdown.
The addition of vitamin E to FtO2B cells in culture clearly has a significant direct protective effect at all concentrations used. Surprisingly it was most effective after an overnight culture and slightly less effective at 7 days. This probably reflects a slower distribution within the cells to where it can exert its beneficial action. For RAW 264.7 cells, however, 5 µM had no protective effect when incubated for 4 hours. After that period, overnight incubation resulted in a dose-dependent tendency for protection against free radicals. After 7 days incubation, protection against free radicals was effectively the same as for the FtO2B cells. These findings correlate well with those observed by Jordao and co-workers in rats [26], but not in patients with alcoholic cirrhosis [5].
Because of the time lag for maximum protection in the FtO2B cells, trolox was substituted for α-tocopherol in one set of experiments with FtO2B and RAW 264.7 cells. Trolox (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid) is a water-soluble vitamin E mimetic that will nevertheless partition into liposomes [44] and has been used as a commercial standard for antioxidant activity. Unlike α-tocopherol, it had no protective effect at concentrations from 10-250 µM when incubated with FtO2B and RAW 264.7 cells for 4 hours in the presence of ethanol. Overnight culture resulted in some protection for FtO2B cells at concentrations of 10 and 50 µM, but pro-oxidant activity for the RAW 264.7 cells at the same concentrations. Why this occurred is not clear, but it is known that trolox can exhibit pro-oxidant activity in the presence of transition metals [45] and free iron may be released from the cells to generate a Fenton-type reaction. Cell cultures of tumorigenic cells have also shown minimal protection against ROS and impaired cell survival [46].
From these data it is clear that protection from free radicals in alcoholic liver disease is dependent not only on the antioxidant, but also the cell type. Naturally occurring vitamin E is a composite of 4 tocopherols and 4 tocotrienols, of which a-tocopherol is the most abundant. Based on our data, it is possible that one of the other 7 compounds may prove to be equally effective, if not more so. Furthermore, cell differences in handling of vitamin E may make true protection problematic unless it can be targeted. Further research is clearly needed into these findings from the above research.
REFERENCES
- Lieber CS (1997) Role of oxidative stress and antioxidant therapy in alcoholic and nonalcoholic liver diseases. Adv Pharmacol 38: 601-628.
- Cederbaum AI, Lu Y, Wu D (2009) Role of oxidative stress in alcohol-induced liver injury. Arch Toxicol 83: 519-548.
- Obih PO, Soblosky JS, Potter BJ (2015) Does ethanol play a pro-oxidant role during oxidative stress in the liver? J Biosciences Meds 3: 1-9.
- Feierman DE, Winston GW, Cederbaum AI (1985) Ethanol oxidation by hydroxyl radicals: role of iron chelates, superoxide, and hydrogen peroxide. Alcohol Clin Exp Res 9: 95-102.
- de la Maza MP, Petermann M, Bunout D, Hirsch S (1995) Effects of long-term vitamin E supplementation in alcoholic cirrhotics. J Am Coll Nutr 14: 192-196.
- Silva CS, Monteiro TH, Simões-Ambrósio LM, Sunaga DY, Cardoso JF, et al. (2013) Effects of α-tocopherol supplementation on liver of rats chronically exposed to ethanol. J Nutrigenet Nutrigenomics 6: 125-136.
- Bhardwaj P, Madan K, Thareja S, Joshi YK, Saraya A (2008) Comparative redox status in alcoholic liver disease and nonalcoholic fatty liver disease. Hepatol Int 2: 202-208.
- Masalkar PD, Abhang SA (2005) Oxidative stress and antioxidant status in patients with alcoholic liver disease. Clin Chim Acta 355: 61-65.
- Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C (2015) Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Sao Paulo Med J 133: 164-165.
- Odeleye OE, Eskelson CD, Watson RR, Mufti SI, Chvapil M (1991) Vitamin E reduction of lipid peroxidation products in rats fed cod liver oil and ethanol. Alcohol 8: 273-277.
- Tsai JH, Chen HW, Chen YW, Liu JY, Lii CK (2010) The protection of hepatocyte cells from the effects of oxidative stress by treatment with vitamin E in conjunction with DTT. J Biomed Biotechnol 2010: 486267.
- Kaur J, Shalini S, Bansal MP (2010) Influence of vitamin E on alcohol-induced changes in antioxidant defenses in mice liver. Toxicol Mech Methods 20: 82-89.
- Lee SJ, Kim SY, Min H (2013) Effects of vitamin C and E supplementation on oxidative stress and liver toxicity in rats fed a low-fat ethanol diet. Nutr Res Pract 7: 109-114.
- Prathibha P, Rejitha S, Harikrishnan R, Das SS, Abhilash PA, et al. (2013) Additive effect of alpha-tocopherol and ascorbic acid in combating ethanol-induced hepatic fibrosis. Redox Rep 18: 36-46.
- Masaki N, Kyle ME, Farber JL (1989) tert-butyl hydroperoxide kills cultured hepatocytes by peroxidizing membrane lipids. Arch Biochem Biophys 269: 390-399.
- Steinbrecher UP (1997) Dietary antioxidants and cardioprotection--fact or fallacy? Can J Physiol Pharmacol 75: 228-233.
- Manson JE, Bassuk SS, Stampfer MJ (2003) Does vitamin E supplementation prevent cardiovascular events? J Womens Health (Larchmt) 12: 123-136.
- Das S, Nesaretnam K, Das DK (2007) Tocotrienols in cardioprotection. Vitam Horm 76: 419-433.
- Mohanty IR, Arya DS, Dinda A, Gupta SK (2013) Comparative cardioprotective effects and mechanisms of vitamin E and lisinopril against ischemic reperfusion induced cardiac toxicity. Environ Toxicol Pharmacol 35: 207-217.
- Rinella ME (2015) Nonalcoholic fatty liver disease: a systematic review. JAMA 313: 2263-2273.
- Ersöz G, Gün?ar F, Karasu Z, Akay S, Batur Y, et al. (2005) Management of fatty liver disease with vitamin E and C compared to ursodeoxycholic acid treatment. Turk J Gastroenterol 16: 124-128.
- Farias MS, Budni P, Ribeiro CM, Parisotto EB, Santos CE, et al. (2012) Antioxidant supplementation attenuates oxidative stress in chronic hepatitis C patients. Gastroenterol Hepatol 35: 386-394.
- Bunchorntavaku C, Wootthananont T, Atsawarungruangkit A (2014) Effects of vitamin E on chronic hepatitis C genotype 3: a randomized, double-blind, placebo-controlled study. J Med Assoc Thai 97: 31-40.
- Lai GY, Weinstein SJ, Taylor PR, McGlynn KA, Virtamo J, et al. (2014) Effects of α-tocopherol and β-carotene supplementation on liver cancer incidence and chronic liver disease mortality in the ATBC study. Br J Cancer 111: 2220-2223.
- Yang M, Gong S, Ye SQ, Lyman B, Geng L, et al. (2014) Non-alcoholic fatty liver disease in children: focus on nutritional interventions. Nutrients 6: 4691-4705.
- Jordão AA Jr, Chiarello PG, Arantes MR, Meirelles MS, Vannucchi H (2004) Effect of an acute dose of ethanol on lipid peroxidation in rats: action of vitamin E. Food Chem Toxicol 42: 459-464.
- Ozdil S, Bolkent S, Yanardag R, Arda-Pirincci P (2004) Protective effects of ascorbic acid, dl-alpha-tocopherol acetate, and sodium selenate on ethanol-induced liver damage of rats. Biol Trace Elem Res 97: 149-162.
- Tsukamoto H, Machida K, Dynnyk A, Mkrtchyan H (2009) “Second hit” models of alcoholic liver disease. Semin Liver Dis 29: 178-187.
- Rosen GM, Rauckman EJ (1981) Spin trapping of free radicals during hepatic microsomal lipid peroxidation. Proc Natl Acad Sci USA 78: 7346-7349.
- Saitoh S, Zhang C, Tune JD, Potter B, Kiyooka T, et al. (2006) Hydrogen peroxide: a feed-forward dilator that couples myocardial metabolism to coronary blood flow. Arterioscler Thromb Vasc Biol 26: 2614-2621.
- Cederbaum AI, Dicker E (1981) Effect of cyanamide on the metabolism of ethanol and acetaldehyde and on gluconeogenesis by isolated rat hepatocytes. Biochem Pharmacol 30: 3079-3088.
- Casavant MJ (2001) Fomepizole in the treatment of poisoning. Pediatrics 107: 170.
- Evans HM, Bishop KS (1922) ON THE EXISTENCE OF A HITHERTO UNRECOGNIZED DIETARY FACTOR ESSENTIAL FOR REPRODUCTION. Science 56: 650-651.
- Brigelius-Flohé R, Traber MG (1999) Vitamin E: function and metabolism. FASEB J 13: 1145-1155.
- Ham AJ, Liebler DC (1997) Antioxidant reactions of vitamin E in the perfused rat liver: product distribution and effect of dietary vitamin E supplementation. Arch Biochem Biophys 339: 157-164.
- Wu D, Cederbaum AI (2009) Oxidative stress and alcoholic liver disease. Semin Liver Dis 29: 141-154.
- von Herbay A, de Groot H, Hegi U, Stremmel W, Strohmeyer G, et al. (1994) Low vitamin E content in plasma of patients with alcoholic liver disease, hemochromatosis and Wilson’s disease. J Hepatol 20: 41-46.
- Bergheim I, Parlesak A, Dierks C, Bode JC, Bode C (2003) Nutritional deficiencies in German middle-class male alcohol consumers: relation to dietary intake and severity of liver disease. Eur J Clin Nutr 57: 431-438.
- Girre C, Hispard E, Therond P, Guedj S, Bourdon R, et al. (1990) Effect of abstinence from alcohol on the depression of glutathione peroxidase activity and selenium and vitamin E levels in chronic alcoholic patients. Alcohol Clin Exp Res 14: 909-912.
- Zhang FK, Zhang JY, Jia JD (2005) Treatment of patients with alcoholic liver disease. Hepatobiliary Pancreat Dis Int 4: 12-17.
- Hartley DP, Petersen DR (1997) Co-metabolism of ethanol, ethanol-derived acetaldehyde, and 4-hydroxynonenal in isolated rat hepatocytes. Alcohol Clin Exp Res 21: 298-304.
- Roy A, Sil PC (2012) Tertiary butyl hydroperoxide induced oxidative damage in mice erythrocytes: Protection by taurine. Pathophysiology 19: 137-148.
- Ochi T (1990) Effects of an organic hydroperoxide on the activity of antioxidant enzymes in cultured mammalian cells. Toxicology 61: 229-239.
- Barclay LR, Artz JD, Mowat JJ (1995) Partitioning and antioxidant action of the water-soluble antioxidant, Trolox, between the aqueous and lipid phases of phosphatidylcholine membranes: 14C tracer and product studies. Biochim Biophys Acta 1237: 77-85.
- Poljsak B, Raspor P (2008) The antioxidant and pro-oxidant activity of vitamin C and trolox in vitro: a comparative study. J Appl Toxicol 28: 183-188.
- Monticone M, Taherian R, Stigliani S, Carra E, Monteghirfo S, et al. (2014) NAC, tiron and Trolox impair survival of cell cultures containing glioblastoma tumorigenic initiating cells by inhibition of cell cycle progression. PLoS One 9: 90085.
Citation: Obih PO, Soblosky J, Potter BJ (2015) Does Vitamin E Protect Against Hepatic Oxidative Stress During Alcohol Metabolism in Rodent Liver Cell Lines? - An EPR Study. J Alcohol Drug Depend Subst Abus 1: 002.
Copyright: © 2015 Obih P, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Journal Highlights
© 2026, Copyrights Herald Scholarly Open Access. All Rights Reserved!