Effect of No Donors on Cytotoxicity in Response to Oxidant Stress
*Corresponding Author(s):
Obih POCollege Of Pharmacy, Xavier University Of Louisiana, New Orleans, Louisiana, United States
Tel:+1 337-482-1000,
Email:poobih@xula.edu
Abstract
The action of nitric oxide (NO) during oxidative stress associated with hepatic alcohol metabolism is controversial. Experiments were designed to examine the effects of exogenous nitric oxide (NO) on the liver cells response to oxidative stress during ethanol metabolism using a rat hepatoma cell line model (Ft02B cells). Oxidative stress was generated by the addition of tert-butyl hydroperoxide (t-BH), a known biological oxidizing agent. FtO2B cells were cultured in the presence or absence of 100 mM EtOH for 3 hours with a range of t-BH concentrations. Cell viability was determined by assaying the culture supernatant for lactate dehydrogenase. In parallel experiments, the effect of an NO donor on Ft02B cells was studied. Similar experiments were carried out with NO inhibitor like NG-nitro-L-arginine methyl ester (L-NAME), NAP, and Disulfiram, an acetaldehyde dehydrogenase inhibitor. In these studies, 400µM t-BH increased cell killing from ca. 4% in medium alone to 29% in the presence of 100 mM ethanol and 500 µM t-BH from 29% to 54%. The addition of an NO donor, S-Nitroso-N-Acetylpenicillamine (SNAP) reduced cell killing in a dose-dependent fashion with 200 uM SNAP having 100% inhibition with ethanol and 400 uM t-BH (4% killing). Adding the NO blocker (L-NAME) to the system resulted in a dose – dependent increase in cell killing (almost 40% at 100 mM L-NAME). Finally, the addition of Disulfiram to this system resulted in a significant increase in cell killing. These results suggest that nitric oxide has a significant protective effect against hepatic oxidative stress during alcohol metabolism.
Keywords
Ethanol; FTO2B cells; Nitric oxide; SNAP; t-BH
INTRODUCTION
Excessive alcohol consumption is the third leading cause of preventable death in the United States and is a risk factor for many health and societal problems. In 2010, the estimated economic cost of excessive drinking in the U. S. was $249 billion [1]. Alcohol enhances the risk for developing several serious medical conditions related to immune system dysfunction, including acute respiratory distress syndrome (ARDS), liver cancer, and alcoholic liver disease (ALD) [2]. In the respiratory system, NO derived from the constitutive type of NO synthase (cNOS, NOS1, NOS3) induces bronchodilation and pulmonary vasodilatation to maintain homeostasis. In contrast, the roles of excessive NO derived from the inducible type of NOS (iNOS, NOS2) in airway and lung inflammation in inflammatory lung diseases including bronchial asthma and chronic obstructive pulmonary disease (COPD) are controversial [3]. The role of NO in alcoholic liver disease is unclear and controversial. For example: “Nitric oxide has pleiotropic effects that obscure whether it predominantly plays a protecting or a damaging role in ALD” [4]. It is also complicated by the interactions of the major cell types within the liver [5]. NO appears to have both pro- and anti-oxidant functions in normal hepatocytes [6]. Nitric oxide regulates a large number of hepatocyte genes [7] and has been suggested to both prevent and potentiate apoptosis [8, 9] Many of the associated proteins are involved in the glutathione anti-oxidant system [10,11], with others from the Caspase family [12] and are affected by S-nitrosylation of cysteine residues. Also affected by S-nitrosylation is alcohol dehydrogenase which has many consequences in alcoholic liver injury, especially since Clemens and co-workers have shown a relationship between acetaldehyde levels and cell survival [13,14]. However, NO also has a greater inhibitory effect on mitochondrial respiration during chronic alcohol consumption [15]. In experimental animals, the results are conflicting: nitric oxide appears to protect from liver injury when produced by non-parenchymal cells, but potentiates it when produced by hepatocytes [16,17] although this may also reflect the lipid content of the two diets used. In the liver, many cell types, including Kupffer cells and hepatocytes have the ability to synthesize NO during endotoxemia and inflammation [18]. RAW 264.7 macrophages and peritoneal macrophages are able to down regulate the ability of hepatocytes to generate nitric oxide [19]. The effects of NO on the liver may also be complicated by its effects on the micro vascular, particularly during alcohol metabolism with associated endotoxemia [20] which could also potentially alter oxygen availability and the hepatic redox status.
The role of Nitric Oxide in alcoholic liver disease is unclear. Some authors have shown that depending on conditions, NO is Janus-faced, in that it may be hepatoprotective or potentiate liver injury depending on both alcohol levels and the extent of prior liver damage [21]. It has also been suggested that eNOS-derived NO is protective against liver disease, but that derived from iNOS activation is deleterious [22]. Nitric oxide, measured as nitrate, has been shown to increase with the progression of chronic liver disease [23]. Since the effects of both ethanol and nitric oxide appear to have cell-specific effects and the hepatocyte is the main metabolic site, it was the aim of this study to examine the consequences of interaction of these substances with a tissue culture model of these parenchymal cells employing rat Ft02B cells.
MATERIALS AND METHODS
FtO2B cells were cultured at 370C in Dulbecco’s modified Eagles medium/Ham’s F12 medium (InVitrogen Life Technologies, Carlsbad CA), supplemented with 10% fetal calf serum, 2mM glutamine, 0.2% sodium bicarbonate, 50 IU/ml penicillin-streptomycin and 1µg/ml amphotericin B in the presence of 5%CO2/95% air. Cells were plated at approximately 25-30% confluence (except where noted) and grown to 80-90% confluence before passaging. FtO2B cells were cultured in the presence or absence of 100 mM ethanol and subjected to oxidative stress through the addition of tertiary-butyl hydroperoxide (t-BH). Ft02B cells were incubated in the presence or absence of 100 mM ethanol for 3 hr with a range of t-BH concentrations. In parallel experiments the effects of SNAP, NAP and Disulfiram additions to FtO2B cultures with t-BH in the presence or absence of ethanol were also assayed [24].
Characterization of ethanol metabolism by Ft02B cells
Ft02B cells, derived from a rat hepatoma, (a kind gift from Dr. Jack Lancaster, jr.) were plated at approximately 1/5 confluence in T-75 tissue culture flasks and cultured in the presence or absence of 100 mM ethanol in the culture medium. The medium was changed daily and assayed for ethanol content. Once plates reached 90-95% confluence, the cells were removed using trypsin-EDTA solution and re-plated at 1/5 confluence. Cell counts and viability assessments were performed by standard tissue culture protocols (see methods section).
Nitric oxide generation
Experiments to investigate nitric oxide production in Ft02B cells have been carried out in earlier investigations of the principal investigator into the consequences of administration of a cytokine mixture (CM) to Ft02 B cells in the presence or absence of NG-nitro-L-arginine methyl esterNG-nitro-L-arginine methyl ester (NMA), a nitric oxide synthase inhibitor. In these experiments, NO production was measured by the Griess reaction and, as such, expressed in terms of nitrite and nitrate.
Addition of S-Nitrosylacetylpenicillamine(SNAP)
Cells were treated with 400 mM t-BH plus a range of SNAP concentrations plus EtOH for 3 hours. Cells were also treated with SNAP alone. Cell killing was measured by LDH release into the supernatant.
NAP
For NAP, FT02B cells were grown to confluency in 12-well plates. Cells were treated for
3 hours with 400 μM of t-BH plus 200 μM NAP or plus 200 μM SNAP. Cell viability was determined with the supernatant using LDH assay.
Treatment with NG-nitro-L-arginine methyl ester (L-NAME) (NO blocker)
Ft02B cells were grown to confluency and incubated for 3 hours as follows: 1) medium alone, 2) 100 mMEtOH plus t-BH, 3) 100 mMEtOH alone. LDH assay was performed at the end of the experiment to determine cell viability.
Treatment with disulfiram
Disulfiram is an agent that inhibits acetaldehyde dehydrogenase and consequently causing acetaldehyde to accumulate. In this experiment, Ft02B cells were grown to confluency and treated with tBH alone, tBH plus SNAP, tBH plus disulfiram or t-BH plus disulfiram plus SNAP with or without EtOH. Cell viability was determined at the end of the experiment.
Statistical analysis
All experiments were performed using a minimum of 5 flasks per time point/experimental variable and repeated at least 3 times. Data was analyzed by either the Student t- test for paired data, or by Analysis of variance for multiple experimental groups. All of the analyses were performed using a commercially available PC-mounted statistical software package (Sigmastat) and tested for normal distribution prior to comprehensive statistical analysis.
RESULTS
Characterization of ethanol metabolism by Ft02B cells
As can be seen in Figure 1, apart from a lag phase of approximately the first day of culturing the cells in the presence of EtOH, there was no significant difference in rate of doubling of these cells. The values given are for the means for 5 flasks/time point. The standard deviations for these results are extremely tight - less than 5% on every day. During this experiment, the cells remained adherent to the flasks, with less than 1% floating in the medium. Viability estimations also showed minimal cell death in the culture.
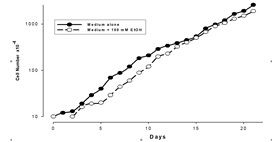 Figure 1: Effect of Ethanol on Ft02B cell growth
Figure 1: Effect of Ethanol on Ft02B cell growth
Dose response curve of t-BH
Ft02B cells were incubated in the presence or absence of 100 mMEtOH for 3 hour with a range of t-BH concentrations. Cell viability was determined by assaying the culture supernatant for lactate dehydrogenase. As may be seen in Figure 2, there was a progressive cytotoxic effect with increasing concentrations of tBH. This was exacerbated by the presence of ethanol. Based on these dose response curves, all subsequent experiments were carried out using 400 mMtBH.
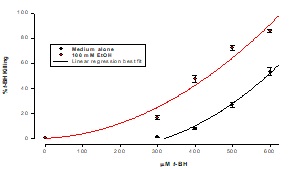 Figure 2: Effect of EtOH on cell killing by t-BH
Figure 2: Effect of EtOH on cell killing by t-BH
Cells were treated with a range of concentrations of t-BH for 3 hours. Killing was measured by LDH release into the supernatant. Results are means ± SD of samples in triplicate.
Addition of SNAP
The addition of SNAP to these cells following the addition of t-BH (an oxidant stress inducer) resulted in a dose-dependent reduction in killing, as may be seen in Figure 3 with 200 µM SNAP having 100% inhibition with 100 mMEtOH and 400 µM t-BH (4% killing) and 65% inhibition with 500 µM t-BH an 100 mM ethanol.
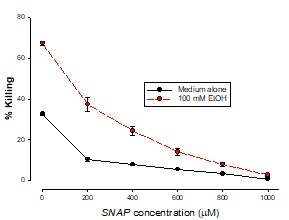 Figure 3: Ambioxidant effect of SNAP with t-BH
Figure 3: Ambioxidant effect of SNAP with t-BH
Cells were treated with 400 mM t-BH plus a range of SNAP concentrations plus EtOH for 3 hours. Cells were also treated with SNAP alone. Killing was measured by LDH release into the supernatant. Results are means ± SD of samples in triplicate.
Addition of NAP
The addition of NOS inhibitor, NAP enhanced the cytotoxic effects of t-BH as may be seen on figure 4.
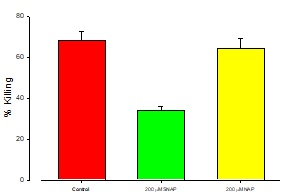
Figure 4: Protective effect of NAP/SNAP on Ft02B cells.
Ft02B cells were treated with 200 μM NAP and 400 μM t-BH or 200 μM SNAP plus 400μM t-BH for 3 hours.
Treatment with L-NAME (NO blocker)
The effect of Ft02B cells treated with L-NAME (NO blocker ) is shown in Figure 5) medium alone, 2) 100 mMEtOH plus t-BH, 3) medium alone, 4) 100 mMEtOH alone. LDH assay was performed at the end of the experiment to determine cell viability. The addition of l-NAME enhanced the cytotoxic effects of t-BH, again in a dose dependent manner. However, as may be seen in figure 5, it had little effect on the cells at low concentration.
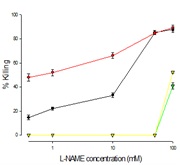 Figure 5: Effect of L-NAME, NO inhibitor on tBH cell killing
Figure 5: Effect of L-NAME, NO inhibitor on tBH cell killing
Cells were treated with a range of L-NAME concentrations with t-BH and EtOH or L-NAME and t-BH without EtOH, or L-NAME alone for 3 hours at the end of which LDH assay was performed.
Treatment with disulfiram
In this experiment, Ft02B cells were grown to confluency and treated with tBH alone, tBH plus SNAP, tBH plus Disulfiram or t-BH plus Disulfiram plus SNAP with or without EtOH. The addition of Disulfiram to the reaction mixture in the presence or absence of SNAP demonstrated that there was a minimal effect of ethanol metabolites in this system, as may be seen in Figure 6.
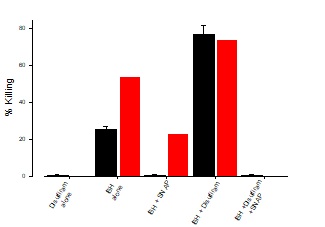 Figure 6: Effect of Disulfiram on t-BH killing
Figure 6: Effect of Disulfiram on t-BH killing
Ft02B cells were treated with media, t-BH alone, t-BH plus SNAP, t-BH plus Disulfiram, or t-BH plus Disulfiram plus SNAP and incubated for 3 hours in the presence or absence of 100 mMetOH. LDH assay was done at the end of the incubation time.
DISCUSSION
Excessive alcohol use, including underage drinking and binge drinking (drinking 5 or more drinks on an occasion for men or 4 or more drinks on an occasion for women), can lead to increased risk of health problems such as injuries, violence, liver diseases, and cancer [25]. Liver disease is the most common medical complication of alcohol abuse; an estimated 15% - 30% of chronic heavy drinkers eventually develop severe liver disease. Alcohol fatty liver is a reversible condition that may progress to alcoholic hepatitis and eventually to cirrhosis and liver failure [26]. Finding ways of limiting excessive alcohol consumption will go a long way to solve this hydra-headed cankerworm. In our studies, the response of added t-BH has previously been shown to be dose-dependent. In these studies, 400 uM t-BH increased cell killing from ca. 4% in medium alone to 29% in the presence of 100 mM ethanol and 500 uM t-BH from 29% to 54%. The addition of an NO donor (SNAP) reduced cell killing in a dose dependent fashion with 200 uM SNAP having 100% inhibition with ethanol and 400 uM t-BH (4% killing) and 65% inhibition with 500 uM t-BH and 100 mM ethanol. The addition of NAP did not affect these values. Adding NO blocker (L-NAME) to the system resulted in a dose–dependent increase in cell killing (almost 40% at 100 mM L-NAME). Finally, the addition of disulfiram to this system resulted in a significant increase in cell killing; but this could be totally inhibited by the presence of 200 uM SNAP (from ca. 70% to 0%).These observations show that nitric oxide is protective against oxidative stress. Some authors have also shown that NO donors acted as antioxidants, preventing lipid peroxidation and restoring GSH levels. L-NAME potentiation of AA-induced toxicity was prevented by the antioxidants Trolox or SAMe. These results demonstrate that NO can be hepatoprotective against CYP2E1- dependent toxicity [27] Some authors have also shown that mitochondrial damage and hypoxia are important features of alcohol-dependent hepatotoxicity and that hepatocytes isolated from chronically ethanol fed animals have decreased cellular bioenergetics reserve capacity which results in increased sensitivity to inhibition by nitric oxide [28]. The same authors also stated that nitric oxide is a major contributor to hypoxic stress in vivo in response to chronic ethanol consumption. NO has been shown to regulate mitochondrial function through its interaction with cytochrome c oxidase, although at higher concentrations, and its combinations with reactive oxygen species, can result in mitochondrial dysfunction [29].Our results suggest that nitric oxide has a significant protective effect against oxidative stress during alcohol metabolism.
REFERENCES
- Sacks JJ, Gonzales KR, Bouchery EE, Tomedi LE, Brewer RD, et al. (2015) 2010 National and state costs of excessive alcohol consumption. American Journal of Preventive Medicine 49:e73-9.
- Curtis BJ, Zahs A, Kovacs EJ (2013) Epigenetic Targets for Reversing Immune Defects Caused by Alcohol Exposure. Alcohol Res 35: 97–113.
- Sugiura H, Chinose M (2011) Nitrative stress in inflammatory lung diseases. Nitric Oxide.25: 138-144.
- Arteel G, Marsano L, Mendez C, Bentley F, McClain CJ, et al. (2003) Advances in alcoholic liver disease. Curr Gastroenterol Rep 17: 625-647.
- Griffon B, Cillard J, Chevanne M, Morel I, Cillard P, et al. (2000) Activated macrophages increase the susceptibility of rat hepatocytes to ethanol-induced oxidative stress: conflicting effects of nitric oxide. Alcohol 35: 230-235.
- Laskin JD, Heck DE, Gardner CR, Laskin DL (2001) Prooxidant and antioxidant functions of nitric oxide in liver toxicity. Antioxid Redox signal 3: 261-271.
- Zamora R, Vodovotz Y, Aulak KS, Kim PKM, Kane JM III, et al. (2002) A DNA microarray study of nitric oxide-induced genes in mouse hepatocytes: implications for hepatic heme oxygenase-1 expression in ischemia/reperfusion. Nitric Oxide 7: 165-186.
- Dambrosio SM, Dambrosio RE, Brady T, Oberyszyn AS, Robertson FM (2001) Mechanism of nitric oxide-induced cytotoxicity in normal human hepatocytes. Environmental and molecular mutagenesis 37: 46-54.
- Kim YM, Talanian RV, Billiar TR (1997) Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J Biol Chem 272: 31138-31148.
- Kurose, I, Higuchi, H, Kato, S, Miura, S, Ishii, H, et al. (1996) Ethanol-induced oxidative stress in the liver. Alcohol Clin Exp Res 20(1 suppl): 77A–85A.
- Corrales FJ, Ruiz F, Mato JM (1999) In vivo regulation by glutathione of methionine adenosyltransferase S-nitrosylation in rat liver. J Hepatol 31: 887-894.
- Li J, Billiar TR, Talanian RV, Kim YM (1997) Nitric oxide reversibly inhibits seven members of the caspase family via S-nitrosylation. BiochemBiophys Res Commun 240: 419-424.
- Clemens DL, Forman A, Jerels TR, Sorrell, MF and Tuma DJ (2002) Relationship between acetaldehyde levels and cell survival in ethanol-metabolizing hepatoma cells. Hepathology 35:1196-1204.
- Clemens, DJ, Sorrell, MF, Tuma, DJ (2003) Ethanol metabolism results in a G2/M cell-cycle arrest in recombinant Hep G2 cells. Hepatology 38: 385–393.
- Venkatraman A, shiva S, Davis AJ, Bailey SM, Brookes PS, et al. (2003) Chronic alcohol consumption increases the sensitivity of rat liver mitochondrial respiration to inhibition by nitric oxide. Hepatology 38: 141-147.
- Baraona E, Zeballos G, Sachet L, Mar KM, Lieber CS (2002) Ethanol consumption increases nitric oxide production in rats, and its peroxynitrite-mediated toxicity is attenuated by polyenylphosphatidylcholine. Alcohol Clin Exp Res 26: 883–889.
- Nanji AA, Greenberg SS, Tahan SR, fogt F, Loscalzo J, et al. (1995) Nitric oxide production in experimental alcoholic liver disease in the rat: role in protection from injury. Gastroenterology 109: 899-907.
- Billiar TR, Curran RD, Stuehr DJ, West MA, Benz BG, et al. (1989) An L-arginine-dependent mechanism mediates Kupffer cell inhibition of hepatocyte protein synthesis in vitro. Journal of Experimental Medicine 169: 1467-1472.
- Griffon B, Cillard J, Chevanne M, More I, Cillard P, et al. (1997) Macrophage-Induced Inhibition of Nitric Oxide Production in Primary Rat Hepatocyte Cultures Via Prostaglandin E2 Release. Hepatology 28: 1300-1308.
- Horie Y, Kimura H, Kato,S, Ohki,E, Tamai,H, et al. (2000) Role of Nitric Oxide in Endotoxin-Induced Hepatic Microvascular Dysfunction in Rats Chronically Fed Ethanol. Alcohol Clin Exp Res 24: 845–851.
- Nanji AA, Jokelainen K, Fotouhinia M, Rahemtulla A, Thomas P, et al. (2001) Increased severity of alcoholic liver injury in female rats: Role of oxidative stress, endotoxin and chemokines. Am J Physiol Gastrointest Liver Physiol 281:G1348-56.
- Iwakiri Y, Kim MY (2015) Nitric oxide in liver diseases. Trends Pharm Sci 36: 524.
- El-Sherif AM, Abou-Shady MA, Al-Bahray AM (2008) Nitric oxide levels in chronic liver disease patients with and without oesophagealvarices. Hepatology International 2: 341-345.
- Obih PO, Potter BJ (2014) Cell-Specific Effects of Ethanol on Nitric Oxide Synthase Induced by Inflammatory Mediators. Pharmacology & Pharmacy 5: 1202-1208.
- Obih PO, Soblosky JS, Potter BJ (2015) Does Ethanol play a Pro-Oxidant Role during Oxidative Stress in the Liver? Journal of Biosciences and Medicines 3: 1-9.
- Center for Disease Control and Prevention (2017). Alcohol and Public Health.
- Masters SB, Trevor AJ (2015) The Alcohols. Basic & Clinical Pharmacology 23: 384-395.
- Wu D, Cederbaum A (2006) Nitric oxide donors prevent while the nitric oxide synthase inhibitor L-NAME increases arachidonic acid plus CYP2E1-dependent toxicity. Toxicol Appl Pharmacol 216: 282–292.
- Zelickson BR, Benavides GA, Johnson MS (2011) Nitric oxide and hypoxia exacerbate alcohol-induced mitochondrial dysfunction in hepatocytes. Biochim Biophys Acta 12: 1573-1582.
Citation: Obih PO, Soblosky J and Potter BJ (2020) Effect of NO Donors on Cytotoxicity in Response to Oxidant Stress. J Alcohol Drug Depend Subst Abus 6: 019.
Copyright: © 2020 Soblosky J, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.