Journal of Nanotechnology Nanomedicine & Nanobiotechnology Category: Medical
Type: Research Article
Electromagnetic Enhancement of Cancer Cells as Biomarkers for Nano Theranostics
*Corresponding Author(s):
Ronald L HustonDepartment Of Mechanical And Materials Engineering, University Of Cincinnati, Cincinnati, OH 45221-0072, United States
Tel:+513 5566133,
Fax:+513 5563390
Email:ron.huston@uc.edu
Received Date: Apr 27, 2017
Accepted Date: Jun 03, 2017
Published Date: Jun 19, 2017
Abstract
“Theranostics” refers to therapy and diagnosis. This paper presents a new method for therapy or diagnosis (or both) for malignant tumors. The underlying premise is that cancer cells with their supernumerary centrioles have a significantly higher electromagnetic field than that of normal cells. This enhanced field can then serves as a “biomarker” or target for the attraction of nanoparticles - specifically: Super Paramagnetic Nanoparticles (SPIONS) which can provide the desired theranostics. Divided into nine sections, the paper provides the bases for these assertions.
INTRODUCTION
Perhaps the greatest challenge for nanotechnologists is the development of nanoparticles for finding and destroying cancer cells. In this paper we review efforts toward this end. We then propose a new method for accomplishing this.
The paper provides an elaboration of a poster presented at Herald International Conference on Nanomedicine & Nanotechnology [1].
The premise is that cancerous tissue has a greater electromagnetic field surrounding it than that of normal tissue. This enhanced field can then be a target biomarker for charged nanoparticles.
As early as 1969 Paul Schafer claimed that disrupted centriole geometry is found in cancer cells [2-4]. He also showed experimentally that large magnetic fields can distort centriole geometry (Centrioles are small perpendicular cylindrical structures lying adjacent to the nucleus in human and animal cells).
Unfortunately Schafer’s findings received relatively little attention since it was apparently overshadowed by the discovery and interpretation by Watson and Krick at about the same time, of the double helix within the cell nucleus [5]. But then, at about the turn of this 21st century, biological researchers began to look again at disrupted centrioles and clusters of centrioles as cancer markers. References 6-20 provide a sampling of these many investigations.
There remain, however, several significant gaps in nanomedicine and cancer therapy. The greatest of these is the absence of a reliable method for safely delivering nanoparticles to cancer cells without harming normal cells. Also the toxicity of nanoparticles remaining in a body has not been fully investigated. Finally, application with brain tumors presents a special challenge due to the blood-brain barrier.
In a series of recent papers AV Singh, et al., summarize current issues in nanomedicine and cancer therapy [21-25]. They also discuss the gaps in the use of nano particles for cancer therapeutics [25-27].
The paper provides an elaboration of a poster presented at Herald International Conference on Nanomedicine & Nanotechnology [1].
The premise is that cancerous tissue has a greater electromagnetic field surrounding it than that of normal tissue. This enhanced field can then be a target biomarker for charged nanoparticles.
As early as 1969 Paul Schafer claimed that disrupted centriole geometry is found in cancer cells [2-4]. He also showed experimentally that large magnetic fields can distort centriole geometry (Centrioles are small perpendicular cylindrical structures lying adjacent to the nucleus in human and animal cells).
Unfortunately Schafer’s findings received relatively little attention since it was apparently overshadowed by the discovery and interpretation by Watson and Krick at about the same time, of the double helix within the cell nucleus [5]. But then, at about the turn of this 21st century, biological researchers began to look again at disrupted centrioles and clusters of centrioles as cancer markers. References 6-20 provide a sampling of these many investigations.
There remain, however, several significant gaps in nanomedicine and cancer therapy. The greatest of these is the absence of a reliable method for safely delivering nanoparticles to cancer cells without harming normal cells. Also the toxicity of nanoparticles remaining in a body has not been fully investigated. Finally, application with brain tumors presents a special challenge due to the blood-brain barrier.
In a series of recent papers AV Singh, et al., summarize current issues in nanomedicine and cancer therapy [21-25]. They also discuss the gaps in the use of nano particles for cancer therapeutics [25-27].
CENTRIOLES
The interior structure of eukaryotic (human and animal) cells is well known. For our purposes, for tumorigenesis, we focus on centrioles - a small pair of perpendicular cylinders adjacent to the nucleus. Errors in centriole geometry are believed to be a source of tumorigenesis [28].
The centrioles are distinctly different from all other organelles and organs:
1) They have precise geometry
2) They have no membrane cover
3) They may be viewed as hollow cylinders with nine radial blades, each having themselves three hollow cylinders known as “microtubules”, forming the centriole circumference
4) Viewed as a cylinder, each centrioles is approximately 400 to 500 nm long and 200 nm in diameter
5) Centrioles occur in pairs (often called the “mother” and “daughter”) with the daughter being perpendicular to the mother and attached to the mother at the base
6) The centrioles lie in a cloud of many proteins which together with the centrioles is known as the “centrosome”
Figure 1 provides a sketch of a typical centriole pair.
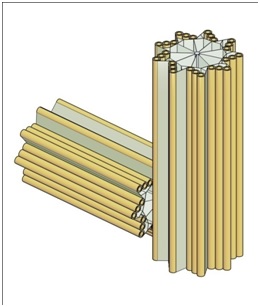
Figure 1: A Typical centriole pair and their centriolar blades.
The centrioles play a central role in cell division. Although this process is generally well understood it may be helpful to some readers to briefly review the cell cycle (birth to separation) [29-33]. The cycle may be viewed in two phases: the “M-phase” (mitosis) and the “interphase” as represented in figure 2.
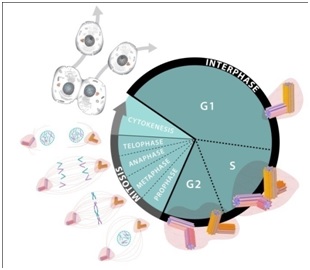
Figure 2: Cell cycle.
The M-phase consists of several subphases called: “Prophase”; “Metaphase”; “Anaphase”; “Telophase”; and “Cytokinesis”. This set of subphases is also known as “mitosis”. Curing mitosis the nucleus is divided and the chromosomes are separated. The cytoplasms with its various organelles are also separated into two halves, each half following one of the nucleus halves.
The Interphase, which is considerably longer than the M-phase also consists of several phases: G1 (or “Gap1”) for growth; S-phase, for more growth and DNA duplication; and G2 (or “Gap2”) for still more growth and for preparation for division.
During the S-phase the centrioles are also duplicated into two pairs, and then during prophase the centriole pairs are separated with the newer pair moving to the opposite side of the nucleus. The centriole pairs now on opposite sides of the nucleus extend their microtubules to form the mitotic spindle, and then pull apart the nucleus [29-33].
As noted previously the centrioles are composed of nine “blades” of Microtubules (MTs) with each blade having three MTs. Thus each centriole has a total of 27 parallel MTs, and some centrioles have an additional two MTs along their axes.
For a typical blade of MTs, each MT does not have the same length: The interior MT (closest to the centriole axis) is the longest. The exterior MT (closest to the centriole perimeter) is the shortest. The third MT then has intermediate length. They are labeled: “A”, “B”, and “C” as represented in figure 3.
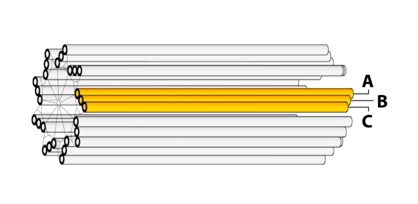
Figure 3: Centriole blades.
New (“daughter”) centrioles are grown from the side at the base of the “mother” centriole, with the axis of the daughter being perpendicular to the axis of the mother. The daughter growth begins on the exterior side of one of the C-MTs.
With there being nine C-MTs, it is not clear which is to be selected for the base of the daughter centriole.
The growing process is believed to occur as follows: During the S-phase of the cell cycle, as the DNA is being separated, a small quantity of Asterless (AsP) is deposed at the base of the selected C-MT. (AsP is the orthologue of Cep152 [34]. The AsP/Cep152 then recruits a patch of the polo-like kinase enzyme: Plk4 which then in turn recruits the protein: SAS-6 for the base of the ensuing daughter centriole.
SAS-6 projects nine outward spokes whose ends attract three spots of Gamma (ϒ) tubulin. The ϒ-Tubulin Ring Complex (ϒTu-RC) and the ϒ-Tubulin Small Complex (ϒTu-SC) [7,35-43].
Figure 4 provides a simplified representation of this process.
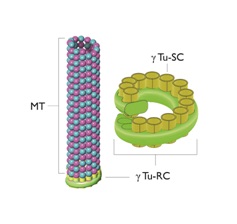
Figure 4: A microtubule, the Gamma-Tubulin Ring Complex (ϒTu-RC) and the Gamma-Tubulin Small Complexes (ϒTu-SCs).
The centrioles are distinctly different from all other organelles and organs:
1) They have precise geometry
2) They have no membrane cover
3) They may be viewed as hollow cylinders with nine radial blades, each having themselves three hollow cylinders known as “microtubules”, forming the centriole circumference
4) Viewed as a cylinder, each centrioles is approximately 400 to 500 nm long and 200 nm in diameter
5) Centrioles occur in pairs (often called the “mother” and “daughter”) with the daughter being perpendicular to the mother and attached to the mother at the base
6) The centrioles lie in a cloud of many proteins which together with the centrioles is known as the “centrosome”
Figure 1 provides a sketch of a typical centriole pair.
Figure 1: A Typical centriole pair and their centriolar blades.
The centrioles play a central role in cell division. Although this process is generally well understood it may be helpful to some readers to briefly review the cell cycle (birth to separation) [29-33]. The cycle may be viewed in two phases: the “M-phase” (mitosis) and the “interphase” as represented in figure 2.
Figure 2: Cell cycle.
The M-phase consists of several subphases called: “Prophase”; “Metaphase”; “Anaphase”; “Telophase”; and “Cytokinesis”. This set of subphases is also known as “mitosis”. Curing mitosis the nucleus is divided and the chromosomes are separated. The cytoplasms with its various organelles are also separated into two halves, each half following one of the nucleus halves.
The Interphase, which is considerably longer than the M-phase also consists of several phases: G1 (or “Gap1”) for growth; S-phase, for more growth and DNA duplication; and G2 (or “Gap2”) for still more growth and for preparation for division.
During the S-phase the centrioles are also duplicated into two pairs, and then during prophase the centriole pairs are separated with the newer pair moving to the opposite side of the nucleus. The centriole pairs now on opposite sides of the nucleus extend their microtubules to form the mitotic spindle, and then pull apart the nucleus [29-33].
As noted previously the centrioles are composed of nine “blades” of Microtubules (MTs) with each blade having three MTs. Thus each centriole has a total of 27 parallel MTs, and some centrioles have an additional two MTs along their axes.
For a typical blade of MTs, each MT does not have the same length: The interior MT (closest to the centriole axis) is the longest. The exterior MT (closest to the centriole perimeter) is the shortest. The third MT then has intermediate length. They are labeled: “A”, “B”, and “C” as represented in figure 3.
Figure 3: Centriole blades.
New (“daughter”) centrioles are grown from the side at the base of the “mother” centriole, with the axis of the daughter being perpendicular to the axis of the mother. The daughter growth begins on the exterior side of one of the C-MTs.
With there being nine C-MTs, it is not clear which is to be selected for the base of the daughter centriole.
The growing process is believed to occur as follows: During the S-phase of the cell cycle, as the DNA is being separated, a small quantity of Asterless (AsP) is deposed at the base of the selected C-MT. (AsP is the orthologue of Cep152 [34]. The AsP/Cep152 then recruits a patch of the polo-like kinase enzyme: Plk4 which then in turn recruits the protein: SAS-6 for the base of the ensuing daughter centriole.
SAS-6 projects nine outward spokes whose ends attract three spots of Gamma (ϒ) tubulin. The ϒ-Tubulin Ring Complex (ϒTu-RC) and the ϒ-Tubulin Small Complex (ϒTu-SC) [7,35-43].
Figure 4 provides a simplified representation of this process.
Figure 4: A microtubule, the Gamma-Tubulin Ring Complex (ϒTu-RC) and the Gamma-Tubulin Small Complexes (ϒTu-SCs).
MICROTUBULES
As noted earlier the centrioles are immersed in a cloud of many proteins with the system of the centrioles and proteins known as the “centrosome”. The centrosome is also known as the Microtubule Organizing Center (MTOC) since the proteins making up the microtubules come out of the centrosome via the ϒTu-SC.
These Microtubules (MTs) are themselves long hollow cylinders - approximately 400 nm long, 25 nm in outside diameter and 15 nm in inside diameter. They are composed of longitudinal connected tubulin dimers. A tubulin dimer is a connected Alpha (α) and Beta (β) tubulin.
Figures 4 and 5 provide sketches of MT and figure 6 shows a sketch of a tubulin dimer.
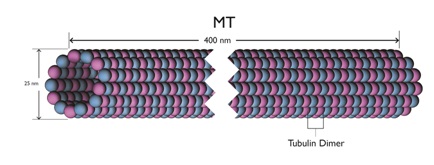
Figure 5: A microtubule length.
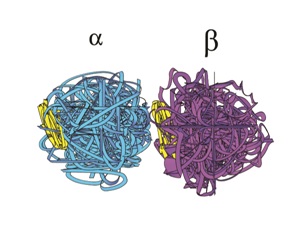
Figure 6: A Tubulin Dimer.
These Microtubules (MTs) are themselves long hollow cylinders - approximately 400 nm long, 25 nm in outside diameter and 15 nm in inside diameter. They are composed of longitudinal connected tubulin dimers. A tubulin dimer is a connected Alpha (α) and Beta (β) tubulin.
Figures 4 and 5 provide sketches of MT and figure 6 shows a sketch of a tubulin dimer.
Figure 5: A microtubule length.
Figure 6: A Tubulin Dimer.
Around the circumference of MT there are 13 filaments. This odd number shows that there is no distinct or uniform pattern of α and β tubulin around the circumference. This in turn implies that the filaments can move longitudinally relative to the overall MT structure.
At the distal end of a MT, if a filament is moving in the distal direction, the end α-β dimers may fall away into the cytoplasm. This activity is known as “catastrophe”, and the length of the MT could be shortened. Alternatively, if the filament is moving in the distal direction and the dimers at the distal end do not fall into the cytoplasm, the MT is lengthened. This is known as “rescue”.
In addition to both catastrophe and rescue, the MT filaments can also oscillate longitudinally.
Reference 44 provides a more detailed review of microtubules.
ELECTROMAGNETICS (EMS)
The α-β tubulin dimers have a negative-positive charge – similar to that of a flashlight battery. A moving charge creates an Electromagnetic (EM) field. A Rapid movement such as a MT filament oscillation can create EM fields. With 27 MTs in each centriole, and with each MT having 13 filaments, there are a total of 351 filaments per centriole, and thus 702 filaments for each centriole pair. The oscillations of this large number of filaments can create strong, varying EM fields.
References 45-49 provide a more detailed review of cellular electromagnetics.
References 45-49 provide a more detailed review of cellular electromagnetics.
DEVELOPMENT OF AN ELECTROMAGNETIC BIOMARKER
In a normal cell there is only one pair of centrioles, except during mitosis when there is two pair. In cancer cells, however, there are numerous (or “supernumerary”) centrioles due to errors in the centriole duplication process [7-20]. While there may be many reasons for the duplication going awry (carcinogens, viruses, radiation) a typical error is a “flowering” centriole as suggested in figure 7.
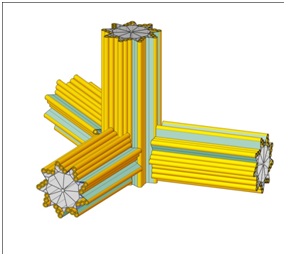
Figure 7: A flowering centriole.
When there are supernumerary centrioles, they tend to cluster together as an enlarged centrosome. This clustering with the resulting thousands of MT filaments will create an enhanced EM field far greater than that of normal cells. This enhanced EM field is then a biomarker distinguishing cancer cells from normal cells. Consequently, this EM biomarker can serve as a target for imaging and therapy. Specifically, in our analysis this EM biomarker is thought to be an ideally suited attractor for magnetically charged nanoparticles.
NANO THERANOSTICS
With theranostics referring to both diagnostics and therapy, if we can get Nanoparticles (NPs) within the cancer cell membrane while at the same time avoiding normal cells, we can both diagnose the extent of the cancerous tumor and also provide therapy to destroy the tumor. Specifically, if the NPs contain metals, such as gold or iron, they can be imaged by x-rays, and thus expose the extent of the tumor. Similarly, if the cell-inserted NPs contain toxic chemicals, the chemical can be released via external control.
If a tumor can be illuminated it can be treated by focused x-rays and/or proton beams. Alternatively, in addition to depositing toxins, magnetically charged NPs can be exposed to oscillating external magnetic fields causing the MTs to vibrate and thermally destroy the cancer cells. In either event the objective is to get NPs inside the cancer cell.
If the NPs are sufficiently small (<20 nm) they will be able to pass through the cell membrane. If the NPs are also magnetic, they will be attracted to the centriole cluster of the cancer cell. The task then becomes: How can we get the NPs sufficiently close to the cancer cells, so the enhanced electromagnetic field of the cancer cell will pull the magnetized NP into the cell? To answer this question, consider first that if the NPs are simply inserted into the blood stream, analogous to chemotherapy, they may be excreted before reaching the tumor or as with chemotherapy the enhanced blood pressure in the vasculature supplying the tumor may tend to push the MTs away from the tumor. Moreover, if the MTs are magnetic, they will be attracted to each other forming clumps which are too large to penetrate the cell membrane, and/or with reduced magnetism to be strongly attracted by the clustered centrioles.
Therefore, what is needed is a direct focus of NPs on the tumor (analogous to the current use of proton beams) but without the NPs clustering together, and also without harm to nearby normal tissue.
To avoid the clustering we can use super paramagnetic NPs which have no magnetic attraction until they are in the presence of a relatively stronger Electromagnetic (EM) field. NPs with iron oxide constituents have been shown to have such properties and are known as “Super paramagnetic Iron Oxide Nanoparticles“(SPIONS) [50-54].
As of this writing the best candidates for development of effective SPIONS are carbon nanotubes with iron oxide cores, known as: “carbon nanoworms”, and similarly carbon nano-spherical shells, known as: “carbon nanospheres”.
If a tumor can be illuminated it can be treated by focused x-rays and/or proton beams. Alternatively, in addition to depositing toxins, magnetically charged NPs can be exposed to oscillating external magnetic fields causing the MTs to vibrate and thermally destroy the cancer cells. In either event the objective is to get NPs inside the cancer cell.
If the NPs are sufficiently small (<20 nm) they will be able to pass through the cell membrane. If the NPs are also magnetic, they will be attracted to the centriole cluster of the cancer cell. The task then becomes: How can we get the NPs sufficiently close to the cancer cells, so the enhanced electromagnetic field of the cancer cell will pull the magnetized NP into the cell? To answer this question, consider first that if the NPs are simply inserted into the blood stream, analogous to chemotherapy, they may be excreted before reaching the tumor or as with chemotherapy the enhanced blood pressure in the vasculature supplying the tumor may tend to push the MTs away from the tumor. Moreover, if the MTs are magnetic, they will be attracted to each other forming clumps which are too large to penetrate the cell membrane, and/or with reduced magnetism to be strongly attracted by the clustered centrioles.
Therefore, what is needed is a direct focus of NPs on the tumor (analogous to the current use of proton beams) but without the NPs clustering together, and also without harm to nearby normal tissue.
To avoid the clustering we can use super paramagnetic NPs which have no magnetic attraction until they are in the presence of a relatively stronger Electromagnetic (EM) field. NPs with iron oxide constituents have been shown to have such properties and are known as “Super paramagnetic Iron Oxide Nanoparticles“(SPIONS) [50-54].
As of this writing the best candidates for development of effective SPIONS are carbon nanotubes with iron oxide cores, known as: “carbon nanoworms”, and similarly carbon nano-spherical shells, known as: “carbon nanospheres”.
DISCUSSION
In addition to SPIONS (e.g., carbon nanoworms and carbon nanopsheres) numerous other nanoparticles have been proposed for tumor treatment, with some success. These include [55-58]:
1) Dendrimers - branching tree-like structures
2) Micelles - spherical, globular structures, with a hydrophobic central core
3) Nanocapsules - fluid filled pouches
4) Fullerenes - carbon bucky balls
5) Liposomes - closed fluid filled spheres with covering layers
6) Polymeric nanoparticles - structures of polymers
7) Ceramic nanoparticles - silicon and other ceramics
8) Metallic nanoparticles - gold, steel, and iron particles
9) Poly(ethylene glycol) nanoparticles - aka “PEGs”
10) SWCNTS - Single-Walled Carbon Nanotubes
Gidwani and Singh have reviewed the possibility and opportunities of using nanoparticles to cross the blood brain barrier [59]. Singh et al., also consider the use of nanoparticles in treating multiple sclerosis and the safety of using carbon nanoparticles in theranostic applications [60,61].
Recently it has been discovered that cancer tumors possess an excess of the vitamin: folic acid [62-65]. Thus, as with an enhanced Electromagnetic (EM) field folic acid is also a biomarker. PEGs (see 9 above) are believed to be attracted to folic acid receptors.
Chemotherapy is a commonly used therapy for cancer. A difficulty with toxic chemicals injected into the blood stream is that the vasculature supplying a tumor is often enlarged with high back pressure. This high pressure can reduce the amount of chemical entering the tumor so that all cells of the tumor are not necessarily reached by the chemical. Over time these unreached cells may cause the tumor to recur. Also, the toxic chemicals are harmful to normal cells.
A principal advantage of nanoparticle therapy is that the nanoparticles can be directed toward the cancer tumor without harmful effects on the surrounding normal tissue with its lower electromagnetic attraction. (Nanoparticles injected into the blood stream are subjected to the same high pressure resistance by the tumor vasculature as the chemical toxins).
Finally although the number of cancer cells in a tumor is large, there is practically no limit to the number of nanoparticles which can be directed toward the tumor’s cancer cells.
The future work is thus well-defined: We need to improve the efficacy of the nanoparticles - particularly the SPIONS and the PEGs.
1) Dendrimers - branching tree-like structures
2) Micelles - spherical, globular structures, with a hydrophobic central core
3) Nanocapsules - fluid filled pouches
4) Fullerenes - carbon bucky balls
5) Liposomes - closed fluid filled spheres with covering layers
6) Polymeric nanoparticles - structures of polymers
7) Ceramic nanoparticles - silicon and other ceramics
8) Metallic nanoparticles - gold, steel, and iron particles
9) Poly(ethylene glycol) nanoparticles - aka “PEGs”
10) SWCNTS - Single-Walled Carbon Nanotubes
Gidwani and Singh have reviewed the possibility and opportunities of using nanoparticles to cross the blood brain barrier [59]. Singh et al., also consider the use of nanoparticles in treating multiple sclerosis and the safety of using carbon nanoparticles in theranostic applications [60,61].
Recently it has been discovered that cancer tumors possess an excess of the vitamin: folic acid [62-65]. Thus, as with an enhanced Electromagnetic (EM) field folic acid is also a biomarker. PEGs (see 9 above) are believed to be attracted to folic acid receptors.
Chemotherapy is a commonly used therapy for cancer. A difficulty with toxic chemicals injected into the blood stream is that the vasculature supplying a tumor is often enlarged with high back pressure. This high pressure can reduce the amount of chemical entering the tumor so that all cells of the tumor are not necessarily reached by the chemical. Over time these unreached cells may cause the tumor to recur. Also, the toxic chemicals are harmful to normal cells.
A principal advantage of nanoparticle therapy is that the nanoparticles can be directed toward the cancer tumor without harmful effects on the surrounding normal tissue with its lower electromagnetic attraction. (Nanoparticles injected into the blood stream are subjected to the same high pressure resistance by the tumor vasculature as the chemical toxins).
Finally although the number of cancer cells in a tumor is large, there is practically no limit to the number of nanoparticles which can be directed toward the tumor’s cancer cells.
The future work is thus well-defined: We need to improve the efficacy of the nanoparticles - particularly the SPIONS and the PEGs.
ACKNOWLEDGEMENT
Support for this research has been provided by the Schafer Foundation for Centriolar Research. The encouragement of Bardyl Tirana and Roger Adelman is also acknowledged, and the assistance of Charlotte Better and Mary Schartman in preparing the manuscript is appreciated. Finally the helpful suggestions of the reviewers are appreciated.
REFERENCES
- Huston RL (2016) Nanoparticle treatment of cancer. Herald’s International Conference and Exhibition on Nanomedicine of Nanotechnology, Baltimore, USA.
- Schafer PW (1969) Centrioles of a human cancer: intercellular order and intracellular disorder. Science 164: 1300-1303.
- Schafer PW, Chandler JA (1970) Electron probe x-ray microanalysis of a normal centriole. Science 170: 1204-1205.
- Schafer PW (1972) Centrioles: intercellular order in normal and malignant cells. J Thorac Cardiovasc Surg 63: 472-477.
- Watson JD, Crick FH (1953) Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 171: 737-738.
- Huston RL (2016) A Review of Centriole Activity, and Wrongful Activity, during Cell Division. Advances in Bioscience and Biotechnology 7: 169-182.
- Nigg EA, Raff JW (2009) Centrioles, centrosomes, and cilia in health and disease. Cell 139: 663-678.
- Schöckel L, Möckel M, Mayer B, Boos D, Stemmann O (2011) Cleavage of cohesin rings coordinates the separation of centrioles and chromatids. Nat Cell Biol 13: 966-972.
- Kobayashi T, Dynlacht BD (2011) Regulating the transition from centriole to basal body. J Cell Biol 193: 435-444.
- Ganem NJ, Godinho SA, Pellman D (2009) A mechanism linking extra centrosomes to chromosomal instability. Nature 460: 278-282.
- Marthiens V, Rujano MA, Pennetier C, Tessier S, Paul-Gilloteaux P, et al. (2013) Centrosome amplification causes microcephaly. Nat Cell Biol 15: 731-740.
- Nigg EA, Stearns T (2011) The centrosome cycle: Centriole biogenesis, duplication and inherent asymmetries. Nat Cell Biol 13: 1154-1160.
- Hatch EM, Kulukian A, Holland AJ, Cleveland DW, Stearns T (2010) Cep152 interacts with Plk4 and is required for centriole duplication. J Cell Biol 191: 721-729.
- Arquint C, Sonnen KF, Stierhof YD, Nigg EA (2012) Cell-cycle-regulated expression of STIL controls centriole number in human cells. J Cell Sci 125: 1342-1352.
- Holland AJ, Lan W, Niessen S, Hoover H, Cleveland DW (2010) Polo-like kinase 4 kinase activity limits centrosome overduplication by autoregulating its own stability. J Cell Biol 188: 191-198.
- Chan JY (2011) A clinical overview of centrosome amplification in human cancers. Int J Biol Sci 7: 1122-1144.
- Bettencourt-Dias M, Hildebrandt F, Pellman D, Woods G, Godinho SA (2011) Centrosomes and cilia in human disease. Trends Genet 27: 307-315.
- Sabino D, Gogendeau D, Gambarotto D, Nano M, Pennetier C, et al. (2015) Moesin is a major regulator of centrosome behavior in epithelial cells with extra centrosomes. Curr Biol 25: 879-889.
- Mahjoub MR, Stearns T (2012) supernumerary centrosomes nucleate extra cilia and compromise primary cilium signaling. Curr Biol 22: 1628-1634.
- Vulprecht J, David A, Tibelius A, Castiel A, Konotop G, et al. (2012) STIL is required for centriole duplication in human cells. J Cell Sci 125: 1353-1362.
- Singh AV (2016) Recent trends in nano-biotechnology reinforcing contemporary pharmaceutical design. Curr Pharm Des 22: 1415-1417.
- Singh AV, Mehta KK (2016) Top-down versus bottom-up nanoengineering routes to design advanced oropharmacological products. Curr Pharm Des 22: 1534-1545.
- Singh AV (2013) Biotechnological applications of supersonic cluster beam-deposited nanostructured thin films: bottom-up engineering to optimize cell-protein-surface interactions. J Biomed Mater Res A 101: 2994-3008.
- Singh AV, Baylan S, Park BW, Richter G, Sitti M (2017) Hydrophobic pinning with copper nanowhiskers leads to bactericidal properties. PLoS One 12: 0175428.
- Singh AV, Sitti M (2016) Patterned and specific attachment of bacteria on biohybrid bacteria-driven microswimmers. Adv Healthc Mater 5: 2325-2331.
- Hassan S, Singh AV (2014) Biophysicochemical perspective of nanoparticle compatibility: a critically ignored parameter in nanomedicine. J Nanosci Nanotechnol 14: 402-414.
- Singh AV, Vyas V, Montani E, Cartelli D, Parazzoli D, et al. (2012) Investigation of in vitro cytotoxicity of the redox state of ionic iron in neuroblastoma cells. J Neurosci Rural Pract 3: 301-310.
- Dart A (2017) Extra! Extra! Read all about it. Nature Reviews Cancer 17: 143.
- Hua VY, Wang WK, Duesberg PH (1997) Dominant transformation by mutated human ras genes in vitro requires more than 100 times higher expression than is observed in cancers. Proc Natl Acad Sci USA 94: 9614-9619.
- Marieb EN (1991) Chapter 3: 600-101 in: Human Anatomy and Physiology (3rdedn), Benjamin/Cummings Publishing, Redwood City, California, USA.
- Guest D (1996) Biology Smart: 133-135. In: The Princeton Review, Random House, New York, USA.
- Hardin J, Bertoni G, Kleinsmith IJ (2017) Becker’s World of the Cell (9thedn). Pearson Education, Washington, USA.
- Campbell NA (1987) Biology. Benjamin Cummings, Redwood City, California, USA. Pg No: 231.
- Galletta BJ, Jacobs KC, Fagerstrom CJ, Rusan NM (2016) Asterless is required for centriole length control and sperm development. J Cell Biol 213: 435-450.
- Pokorný J (2011) Electrodynamic activity of healthy and cancer cells. 19th International Fröhlick’s Symposium J Phys Conf Ser 329: 012007.
- Kollman JM, Polka JK, Zelter A, Davis TN, Agard DA (2010) Microtubule nucleating gamma-TuSC assembles structures with 13-fold microtubule-like symmetry. Nature 466: 879-882.
- Teixidó-Travesa N, Roig J, Lüders J (2012) The where, when and how of microtubule nucleation - one ring to rule them all. J Cell Sci 125: 4445-4456.
- Pokorný J, Hašek J, Jelínek F (2005) Endogenous electric field and organization of living matter. Electromagnetic Biology and Medicine 24: 185-197.
- Strnad P, Gönczy P (2008) Mechanisms of procentriole formation. Trends Cell Biol 18: 389-396.
- Schiebel E (2000) gamma-tubulin complexes: binding to the centrosome, regulation and microtubule nucleation. Curr Opin Cell Biol 12: 113-118.
- Lin TC, Neuner A, Schiebel E (2015) Targeting of γ-tubulin complexes to microtubule organizing centers: conservation and divergence. Trends Cell Biol 25: 296-307.
- Oakley BR, Paolillo V, Zheng Y (2015) γ-Tubulin complexes in microtubule nucleation and beyond. Mol Biol Cell 26: 2957-2962.
- Huston RL (2016) Mechanics of centriole microtubules. Advances in Biosciences and Biotechnology 7: 266-277.
- Huston RL (2016) A Review of Electromagnetic Activity in Cellular Mechanics. Advances in Bioscience and Biotechnology 7: 360-371.
- Cuzick J, Holland R, Barth V, Davies R, Faupel M, et al. (1998) Electropotential measurements as a new diagnostic modality for breast cancer. Lancet 352: 359-363.
- Pokorný J (2012) Physical aspects of biological activity and cancer. AIP Advances 2: 1-11.
- Pokorný J, Pokorný J, Kobilková J (2013) Postulates on electromagnetic activity in biological systems and cancer. Integr Biol (Camb) 5: 1439-1446.
- Pokorný J, Hašek J, Vanis J, Jelínek F (2008) Biophysical aspects of cancer--electromagnetic mechanism. Indian J Exp Biol 46: 310-321.
- Cheeseman IM, Desai A (2008) Molecular architecture of the kinetochore-microtubule interface. Nat Rev Mol Cell Biol 9: 33-46.
- Moghimi SM, Hunter AC, Murray JC (2005) Nanomedicine: current status and future prospects. FASEB J 19: 311-330.
- Jordan A, Scholz R, Maier-Hauff K, van Landeghem FK, Waldoefner N, et al. (2006) The effect of thermotherapy using magnetic nanoparticles on rat malignant glioma. J Neurooncol 78: 7-14.
- Park JH, von Maltzahn G, Zhang L, Derfus AM, Simberg D, et al. (2009) Systematic surface engineering of magnetic nanoworms for in vivo tumor targeting. Small 5: 694-700.
- Haley B, Frenkel E (2008) Nanoparticles for drug delivery in cancer treatment. Urol Oncol 26: 57-64.
- Byrne JD, Betancourt T, Branon-Peppas L (2008) Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv Drug Deliv Rev 60: 1615-1626.
- Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, et al. (2008) Nanoparticles in medicine: therapeutic applications and developments. Clin Pharmacol Ther 83: 761-769.
- Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, et al. (2007) Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol 2: 751-760.
- Gidwani M, Singh AV (2014) Nanoparticle enabled drug delivery across the blood brain barrier: in vivo and in vitro models, opportunities and challenges. Curr Pharm Biotechnol 14: 1201-1212.
- Singh AV, Khare M, Gade WN, Zamboni P (2012) Theranostic implications of nanotechnology in multiple sclerosis: a future perspective. Autoimmune Dis Pg no: 12.
- Singh AV, Mehta KK, Worley K, Dordick JS, Kane RS, et al. (2014) Carbon nanotube-induced loss of multicellular chirality on micropatterned substrate is mediated by oxidative stress. ACS Nano 8: 2196-2205.
- Rosenholm JM, Meinander A, Peuhu E, Niemi R, Eriksson JE, et al. (2009) Targeting of porous hybrid silica nanoparticles to cancer cells. ACS Nano 3: 197-206.
- Lin MM, Kim DK, El Haj AJ, Dobson J (2008) Development of Superparamagnetic Iron Oxide Nanoparticles (SPIONS) for translation to clinical applications. IEEE Trans Nanobioscience 7: 298-305.
- Zhang Y, Kohler N, Zhang M (2002) Surface modification of superparamagnetic magnetite nanoparticles and their intracellular uptake. Biomaterials 23: 1553-1561.
- Kukowska-Latallo JF, Candido KA, Cao Z, Nigavekar SS, Majoros IJ, et al. (2005) Nanoparticle targeting of anticancer drug improves therapeutic response in animal model of human epithelial cancer. Cancer Res 65: 5317-5324.
- Liong M, Lu J, Kovochich M, Xia T, Ruehm SG, et al. (2008) Multifunctional inorganic nanoparticles for imaging, targeting, and drug delivery. ACS Nano 2: 889-896.
Citation: Huston RL (2017) Electromagnetic Enhancement of Cancer Cells as Biomarkers for Nano Theranostics. J Nanotechnol Nanomed Nanobiotechnol 4: 015.
Copyright: © 2017 Ronald L Huston, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Journal Highlights
© 2026, Copyrights Herald Scholarly Open Access. All Rights Reserved!