Epithelial To Mesenchymal Transition in Breast Cancer Metastasis: Mitochondria Take the Center Stage
*Corresponding Author(s):
Manti GuhaDepartment Of Biomedical Sciences, School Of Veterinary Medicine, University Of Pennsylvania, Philadelphia, United States
Tel:+1 2158986830,
Email:manti@upenn.edu
Abstract
Epithelial to Mesenchymal Transition (EMT), a process involved in organogenesis and wound healing is now at the center stage of cancer metastasis. While most of the current research is focused on defects arising in the nuclear genome, the contribution of defects in mitochondria has remained under investigated. In the past decade, the association between mitochondrial genomic (mtDNA) and functional defects resulting in altered metabolism with tumor progression and poor outcome has become more evident. Here we review the contribution of mitochondria in epithelial-to-mesenchymal transition, particularly focusing on breast cancer. Our goal is to highlight the critical role that mitochondrial genome defects induced retrograde signaling play in driving cellular plasticity. We will further discuss the molecular intermediates of the retrograde signaling pathway, which can potentially be novel therapeutic targets in mitochondrial stress induced EMT.
Keywords
ABBREVIATIONS
brCSC : breast Cancer Stem Cells
Ca2+ : Calcium
C/EBPδ : CCAAT/Enhancer Binding Protein delta
Cn : Calcineurin
CREB : Camp Response Elements Binding Protein
ECM : Extra Cellular Matrix
EMT : Epithelial To Mesenchymal Transition
ER : Estrogen Receptor
ESRP1 : (Epithelial Splicing Regulatory Protein 1)
FOXC1 : Forkhead Box C1
HIF : hypoxia inducible factor
hnRNPA2 : heterogeneous nuclear Ribonucleoprotein A2
mtDNA : mitochondrial DNA
NFATc : Nuclear Factor of Activated T-cells, Cytoplasmic, Calcineurin-Dependent 1
NFκB : Nuclear Factor kappa-light-chain-enhancer of activated B cells
PR : Progesterone Receptor
rRNA : ribosomal RNA
TFAM : Transcription Factor A, Mitochondrial
TGFβ1 : Transforming Growth Factor beta
tRNA : transfer RNA
EPITHELIAL-TO-MESENCHYMAL TRANSITION (EMT) IN CANCER CELL METASTASIS
Metastasis accounts for over 90% of cancer associated mortalities, despite recent advances in detection and treatment options of cancer patients [1,2]. Metastasis involves a series of processes by which a primary tumor spreads from its initial site to secondary tissues/organs. Over 80% of all carcinomas are epithelial in origin. In order to invade, disseminate to distant tissues and subsequently form metastatic colonies, neoplastic epithelial cells have to acquire, at least transiently, a more mesenchymal phenotype [3,4]. Cellular plasticity during this transition between epithelial and mesenchymal phenotypes is achieved by the activation of a cascade of transcriptional and cell biological events termed the Epithelial-to-Mesenchymal Transition (EMT) [5-9]. During an EMT, tumor epithelial cells lose their epithelial characteristics, including cell-cell adhesion, baso-apical polarity and motility, and acquire mesenchymal traits, including motility, invasiveness and, importantly, many of the attributes of stem cells. The involvement of cellular alterations resembling an EMT in tumor cell metastasis have been well documented in many types of carcinomas, including breast, prostate, colon, head and neck, ovary and lung cancers.
The process of tumor metastasis involves a number of sequential cellular events. The tumor cells disseminate from the primary mass, invade into the surrounding cellular matrix, intravasate into the lymphatic and blood vessels, enter and survive in the circulation, extravasate out from the capillary, home-in at a distant organ and proliferate to form secondary tumors in distant organs [10,11]. Acquired migratory capacity is a prerequisite for the epithelial carcinoma cell within the primary tumor mass to escape from the primary tumor and enter the circulation. Cell migration is achieved by multistep interrelated processes involving the formation of lamellipodia/membrane protrusions at the front edge, cycles of adhesion and detachment, cell body contraction, and tail retraction [12]. Most cells have the intrinsic potential for directional movement involving extension of the leading edge during normal cellular processes such as development, tissue remodeling and regeneration. The migratory pattern of tumor cells is random and involves front-rear polarity. The turnover of focal adhesion factors plays a pivotal role in tumor cell migration. Focal adhesions at the leading edge provide traction points moving the cell body forward while disassembly of focal adhesions at the rear allows the retraction of the rear and results in translocation of the cell in the direction of movement [13]. This migratory pattern is one of the hallmarks of aggressive tumors indicative of the transition from a benign to a malignant phenotype. To extravasate through capillary endothelium, cancer cells need to adhere to endothelial cells. The invasion into the surrounding matrix, requires the disassembly of the adhesions between cancer cells and the extracellular matrix [13]. Infact as high as 20% of the candidate genes involved in breast and colorectal carcinogenesis are adhesion-related genes, implying the significance of cell adhesion in cancer progression [14].
SIGNALING FACTORS INVOLVED IN EPITHELIAL-TO-MESENCHYMAL TRANSITION
A number of signaling factors have been shown to drive EMT both under physiologic and pathologic conditions and these have been discussed in many previous reviews. Therefore in this review, we will briefly discuss the role of three major factors which are “drivers” of several downstream pathways involved in driving EMT: (1) hypoxia, (2) cellular Calcium (Ca2+) levels and (3) metabolic reprogramming.
The hypoxic environment within the core of the solid tumor has been established as one of the crucial cellular factors driving EMT [15-19]. Uncontrolled cell proliferation of tumor cells leads to larger tumor mass. This results in limited availability of nutrients and oxygen in the microenvironment exposing tumor cells to intermittent hypoxic conditions. Higgins et al., demonstrated that hypoxia-induced EMT in renal epithelial cells depends on Hypoxia-Inducible Factors (HIF)-dependent signaling [18]. Studies by Copple et al., also show that HIF-1α is important to induce EMT in hypoxic hepatocytes [20]. Luo et al., provided a strong evidence for the involvement of HIF-1α in inducing EMT by silencing HIF at 2% oxygen and over-expression of an oxygen-insensitive HIF mutant at 21% oxygen. Interestingly, expression of the oxygen-insensitive HIF mutant abrogated HIF mediated Snail activation and subsequent cell migration [21]. Their report identified Snail as a HIF target gene and provides novel insights into the regulation of Snail during hypoxia-induced EMT. In addition to HIF signaling, tumor cells activate transforming growth factor, TGFβ1 in response to hypoxia. The role of TGFβ in EMT has been demonstrated in numerous studies. A more direct evidence comes from a report showing Erbin, a Smad/Erk pathway inhibitor inhibits TGFβ induced EMT in renal tubular epithelial cells [22,23].
Another important factor which has emerged as a critical regulator of cell migration is Ca2+ which is the ubiquitous second messenger [24]. Ca2+ flux has been shown to be important for the migration of various cell types, including tumor cells [25-29]. Store-operated calcium is the predominant Ca2+ entry mechanism particularly in non-excitable cells [30,31]. As mentioned in the earlier section, focal adhesion turnover is critical for tumor cell migration during metastasis. Blocking store-operated Ca2+ influx slows down focal adhesion turnover, resulting in larger focal adhesions and stronger adherence that would impede the fast migration of metastatic tumor cells. Reduction of calcium channel proteins Orai1 or STIM1 either by genetic targeting in highly metastatic human breast cancer cells or treatment with a pharmacological inhibitor of store-operated calcium channels was reported to reduce tumor metastasis in animal models. Therefore, agents that block store-operated Ca2+ channels, such as SKF96365, genetically silencing Orai1 and STIM1, or antibodies that specifically block the channel activity of store-operated calcium could be potential therapeutics for tumor metastasis [32].
A number of reports provide evidence that metabolic reprogramming in tumor cells is a driver of tumor metastasis. The metabolic plasticity of tumor cells has gained renewed interest with the goal to defining novel pathways based on metabolic parameters involved in tumor initiation and progression [33]. Using a high-throughput metabolic phenotyping platform, it has been recently demonstrated that breast cancer stem cells, which are formed during an EMT, have a higher ability to utilize additional catabolic fuels perhaps as an adaptive survival mechanism [34]. This study showed that cancer stem cells acquired nutrients from glycolysis end products (pyruvate and lactate) and ketone bodies (β hydroxybutyrate) from extracellular microenvironment to support their mitochondrial functions. We have earlier reported that a metabolic switch to glycolysis and activation of Insulin-like Growth Factor-1 Receptor (IGF1R)/Akt1 kinase pathway and activation of glucose transporter-4 all of which were critical to the survival of tumorigenic transformation in our cellular model [35-37]. We observed a glycolytic switch accompanied by activation of the Akt1 kinase pathway in breast cancer cells during an EMT induction [38]. Another study reported in two different breast cancer cell lines, HER2-positive BT-474 and MCF-7, EMT induction was accompanied by increased aerobic glycolysis, over expression of glucose transporters, lactate dehydrogenase isoforms, monocarboxylate transporters and glycogen phosphorylase isoform. Interestingly enzymes involved in anabolic pathways and gluconeogenesis were suppressed during EMT. Their observation that the metabolic changes are similar in different breast cancer cells during EMT induction, suggests the generality of the metabolic transition [39]. These differences in the metabolic properties between the cancer stem cells and the primary tumor cells can therefore be harnessed in designing therapeutic strategies targeting cells with metastatic potential.
INVOLVEMENT OF EMT IN BREAST CANCER METASTASIS
In normal and neoplastic epithelial cells in vitro, EMT has been induced by various growth factors, including TGFβ1, hepatocyte growth factor and PDGF. These growth factors and their cognate receptors subsequently activate an array of transcription factors, which have the potential of inducing an EMT [40]. The Notch and β catenin signaling pathway has been established as a major driver of EMT by activating downstream transcription factors in various cancers including breast cancers [41-43]. These factors include the homeobox protein Goosecoid (Gsc), the zinc-finger proteins Snai1 (Snail) and Snai2 (Slug), the basic helix-loop-helix protein Twist1 (Twist), the forkhead box proteins FOXC1 and FOXC2, and the zinc-finger, E-box-binding proteins Zeb1 and Sip1 (Zeb2) [6,44-49]. In addition to activation of transcription factors, members of the miR-200 family of micro-RNAs are down-regulated during an EMT [50-52]. Downregulation of these miRNAs subsequently results in the up-regulated expression of several critical target genes, notably Zeb1 and Zeb2. Therefore expression or activation of any one of these transcription factors or down-regulation of the miR-200 family in neoplastic epithelial cell is sufficient to reprogram these cells into an EMT. Moreover, many of these factors are expressed concomitantly in the mesenchymal cells that have passed through an EMT. A number of comprehensive reviews on the contributions of each inducer to the EMT program have already been published before.
It is becoming evident that understanding the mechanisms and cellular pathways in breast cancer holds the key to improving therapeutic response in clinics. Therefore, studies have focused on identifying the molecular signatures of breast cancer types and signaling pathways that govern prognosis. Based on microarray analyses clinical breast cancers have been classified into a number of distinct subtypes, such as luminal A and B, triple (ER-, PR- and Her2-)-negative (TNBC), and HER2-expressing tumors. Significantly high numbers of TNBC tumors are basal-like in origin [53-60]. These subtypes have been useful in designing therapeutic approach, predicting metastasis and survival. In recent years, additional molecular subtypes of tumors have been characterized based on high throughput genomic and proteomic analyses [55,61]. A recent study proposed a computational method of Weighted Similarity Network Fusion (WSNF) to identify the microRNA-Transcription Factors - messenger RNA regulatory networks [62].
Developing novel computational approaches will be useful to characterize the functional relevance of the genomic drivers of breast tumor metastasis [63]. Several studies have identified individual factors that induce EMT in breast cancer. Two independent studies aimed to identify an EMT core gene signature either by ectopically expressing Gsc, Snail, Twist, or TGFβ1, or by silencing E-cadherin. These studies demonstrated that gene expression changes associated with the above mentioned EMT-inducing transcription factors correlated most closely with the claudin-low and metaplastic breast cancers [46,64]. Analysis of mRNA expression levels in cells over expressing EMT-inducers revealed that Twist, Snail, and TGFβ1 upregulate expression of Foxc2, Zeb1, and Zeb2. In a study designed to identify which of the established EMT inducers had the strongest correlation with poor prognostic outcome, it was shown that FOXC1 expression most closely correlated with a poorer survival of the breast cancer patients in the NKI and UNC datasets [46]. Therefore, FOXC1 is a potential strong drug target in metastatic breast cancer. Notably FOXC1 is highly expressed in metaplastic and basal-like breast cancer subtypes for which highly effective treatments are not currently available.
INVOLVEMENT OF MITOCHONDRIAL DEFECTS IN CANCER AND EMT
Mitochondria, the cellular energy house which generates ATP, have emerged in the past decade as major cell signaling hubs regulating cell death and cell proliferation. Mitochondrial biogenesis and functions are controlled by both the nuclear and mitochondrial genome. Each cell contains 100-1000s of mitochondria depending on the energy requirement of the tissue and each mitochondrion contains several copies of mitochondrial genome (mtDNA). Human mtDNA encodes 13 essential subunits of the Oxidative Phosphorylation (OXPHOS) system as well as 2 rRNAs and 22 tRNAs used in mitochondrial translation. MtDNA is highly susceptible to damage from numerous cellular factors including reactive free radicals produced by electron transport chain, the hypoxic environment within solid tumors, defects in mtDNA transcriptional machinery and environmental toxins. Ironically many chemotherapeutic drugs targeting primary tumors damage mtDNA and impair mitochondrial functions. Furthermore, reduction in functional mtDNA copies by increase in ratios of mutant: wild-type mtDNA owing to time-dependent accumulation of mutant DNA or reduction of mtDNA copy number resulting from defective replication are hallmarks of many pathological conditions. Loss of functional mtDNA resulting from mutations reported in nuclear encoded factors which control mtDNA transcription and translation such as polymerase gamma, the mtDNA helicase TWINKLE and defective TFAM [65,66]. Normal mitochondrial functions can be adversely affected by both mutations and mtDNA polymorphisms.
Impaired oxidative phosphorylation, a consequence of mitochondrial dysfunction, has been shown to be involved in tumorigenesis. One of the early theories about the involvement of mitochondrial dysfunction in tumors was derived from Warburg’s hypothesis. Warburg observed that cancer cells were high in fermentation and low in respiration which led to his hypothesis that tumor cells originated from non-neoplastic cells which adopted anaerobic metabolism as an adaptive mechanism after defects in its respiratory system [67]. Even though Warburg’s hypothesis remains contentious, changes in the number, shape, and impaired mitochondrial functions have been reported in various cancers in agreement with Warburg’s hypothesis [68,69]. Interestingly, abnormal mtDNA was observed in leukemic myeloid cells by electron microscope [70,71]. In the past decades, mutations in both the non-coding and coding regions of the mtDNA have been identified in various types of cancer.
Defects in mitochondrial enzymes, Succinate Dehydrogenase (SDH) [72-84] fumarate hydratase (fumarase) [85-91] (IDH) [92-108] account for deregulated bioenergetics and tumor cells’ mitochondrial dysfunction favoring tumor progression. Moreover accumulation of these metabolites has been shown to cause epigenetic alterations by influencing the activities of histone and DNA methylases [109-111]. Nuclear -encoded mitochondrial deacetylase SIRT3 has also been shown to play a role as a tumor suppressor and loss of SIRT3 has been shown to increase tumorigenicity in various cancers [112-117]. Altered epigenetic status is associated with acquired stemness in cancer cells therefore it is of importance that changes in metabolism and mitochondrial functions can affect epigenetics driving an EMT phenotype.
A number of studies have shown that defects in nuclear DNA encoding mitochondrial proteins have been reported to be involved in tumorigenesis via either increasing production of Reactive Oxygen Species (ROS) or activation of Ca2+ dependent signaling [37,118-122]. As mentioned earlier, the elevated levels of reactive free radicals in the hypoxic core of the solid tumors impairs mitochondrial functions. Dysfunctional electron transport chain in turn results in increased production of mitochondrial ROS and the ROS dependent signaling which determine tumor cell fate [123]. Moreover ROS or Nuclear Factor kappa B (NFκB) have been reported to facilitate EMT in certain cell types and in a TNF α- dependent manner [124]. Interestingly, it has been demonstrated that H2O2 alone can promote EMT, mediated by NFκB, independent from TNFα-induced signaling. In numerous cell lines and animal models, EMT has been reported to be induced by various signaling pathways and numerous transcription factors [125-127] and mitochondrial ROS induced signaling pathway may be one of the leading factors of EMT. The contribution of mitochondrial stress induced Ca2+ signaling is discussed in a later section.
MITOCHONDRIAL DEFECTS IN BREAST CANCER
Significant progress has been made in understanding the contribution of defects in the nuclear genome towards breast cancer metastasis. However, the mitochondrial heterogeneity among breast cancer patients has remained a neglected area of breast cancer research. Defects in mitochondrial functions, mitochondrial genome defects including reduced copy numbers, germline or somatic mtDNA mutations and Microsatellite Instability (MSI) have been reported in a high percentage of breast cancer patients [128-134]. Clinical reports from various cohorts and The Cancer Genome Atlas (TCGA) show a significant percentage of hormone receptor (Estrogen and Progesterone receptor) and Herceptin (HER2) Triple Negative Breast Tumors (TNBC) are highly aggressive and have poor prognostic outcomes. Tumors with reduced mtDNA copy numbers correlate with high metastasis and poor 5 year disease free survival rates in the absence of neoadjuvant chemotherapy [135]. A number of studies have identified the D-Loop of the mtDNA as the “hot spot” for mutations associated with over 42% breast cancers [136,137]. The G10398A mtDNA polymorphism was the one of the early reported associations with an increased risk of breast cancer in African-American women [138]. One of the most frequently observed mtDNA defect in breast tumors in the ?mtDNA4977 deletion [133,139-142]. Other frequently reported mtDNA mutations associated with breast cancer is at the NADH dehydrogenase subunits 1,2,4,5 ATP synthase F0 subunit 8, cytochrome c oxidase subunit III. Mutations in 12s and 16s rRNA have also been reported in breast cancer patients [132,143]. Another study identified novel tRNA mutations in two breast cancer cell lines [144]. It has been shown that introducing mitochondria from highly invasive breast cancer cell lines into noninvasive cells imparted increased tumorigenicity in the recipient cell [144,145]. It is now broadly accepted that mitochondria are significant contributors towards the cellular tumorigenic transformation.
MITOCHONDRIAL RETROGRADE SIGNALING IN EMT
In the last decade, cellular signaling originating from dysfunctional mitochondria has been implicated in tumorigenic transformation in various cancers. Based on studies from various independent laboratories, broadly, these can be categorized into three different pathways depending on the stress inducer and/or propagator of the signaling: (1) Ca2+ activated calcineurin mediated signaling; (2) Mitochondrial reactive oxygen species dependent signaling; (3) Mitochondrial unfolded protein (UPRmt) induced signaling.
The observation that dysfunctional mitochondria activate a cytosolic Ca2+ / calcineurin dependent signaling was initially made by the Avadhani laboratory in 1999 and is one of the most well-defined mitochondrial stress signaling pathways. Mitochondrial stress, owing to either reduction in mtDNA copy number (70-80% depletion) or defects in electron transport chain complexes initiates a mitochondrial retrograde signaling pathway (Figure 1) [35-37,145-155]. A consequence of impaired electron chain complex is the disruption of mitochondrial membrane potential. Perturbation of mitochondrial membrane potential (??m) affects the mitochondrial calcium uptake leading to increased cytosolic calcium. It is interesting that the onset of mitochondrial stress signaling involves calcium, which as mentioned earlier is a regulator of cell motility suggesting that mitochondrial retrograde signaling confers migratory capacity. This activates a Ca2+ dependent phosphatase, Calcineurin (Cn) and a set of nuclear transcription factors, such as CREB, C/EBPδ, NFATc, NFκB (cRel:p50) and heterogeneous Ribonucleoprotein A2 (hnRNPA2). The read out of this signaling is an alteration in the expression profile of nuclear genes involved in various oncogenic pathways [152-154].

Figure 1: Overview of EMT induced by partial depletion of mtDNA in mammary epithelial cells by activation of retrograde signaling. Pink: epithelial cells, Blue: carcinoma cells, Green: epithelial cells transitioned to mesenchymal cells.
While reduction in mtDNA has been reported in 63-80% of breast tumors, neither the functional relevance of this correlation nor the underlying mechanisms by which mtDNA reduction induces EMT was investigated until recently. We reported that 70-80% reduction in mtDNA copy number and impaired cellular respiration in both breast cancer MCF7 and normal MCF10A cells results in an epithelial-to-mesenchymal transition (Figure 1) characterized by loss of cell polarity and cell-ECM adhesions with an increase in migration, 3D tumorsphere-formation (Figures 2 and 3) and acquired invasive phenotype [38]. Interestingly, we observed the activation of calcineurin which is a propagator of the mitochondrial retrograde signaling. Downstream of activation of calcineurin, mesenchymal genes were induced along with a loss of epithelial markers conforming with the morphological transition from epithelial to mesenchymal state. The contribution of reduced mtDNA induced signaling in driving this cellular plasticity was evident by restoring the mtDNA contents which reversed the transition to epithelial state. A recent proteomic analyses aimed at identifying the altered proteins involved in metastasis of TNBCs showed that Single-Strand Binding Protein (SSBP-1) was downregulated in metastatic cells [156]. Decreased SSBP-1 expression resulted in loss of mtDNA copy number [156] and triggered a calcineurin mediated mitochondrial retrograde signaling similar to that previously defined from our group [35-37,148,149,152-154,157].
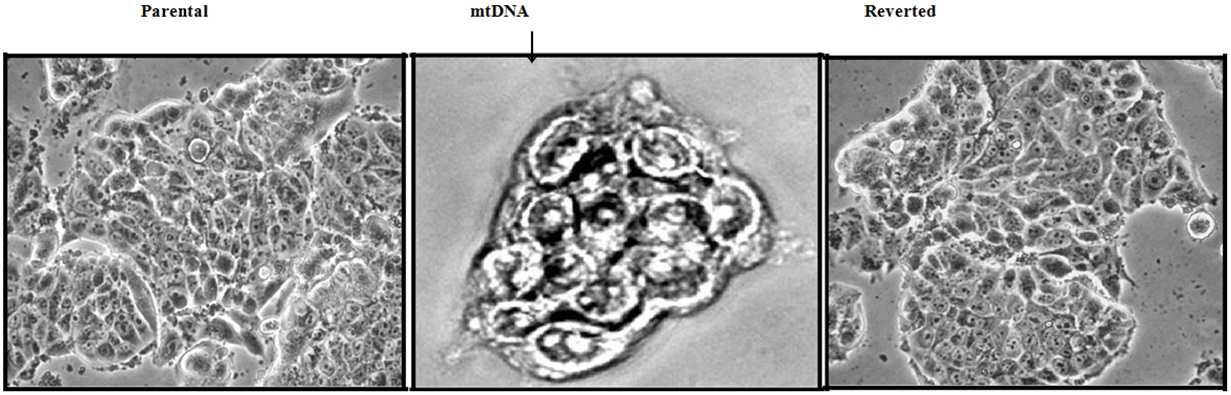
Figure 2: Bright Field images (20x magnification) of two dimensional cultures of showing altered morphology between parental, mtDNA-reduced and mtDNA-rescued (reverted) MCF7cells. MtDNA-reduced MCF7 cells showed loss of contact inhibition and formed piled up colonies.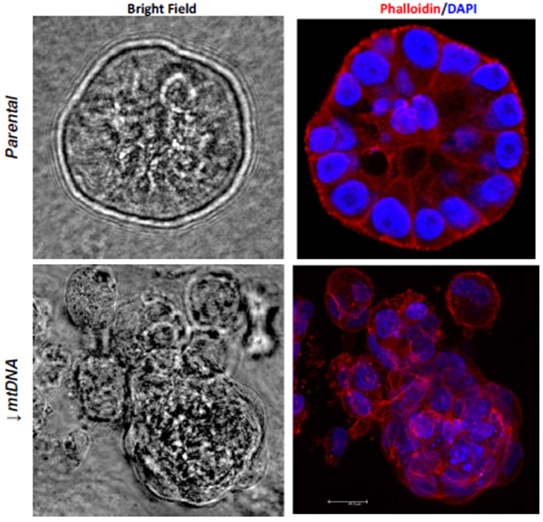
Figure 3: Three dimensional mamospheres formed by parental and mtDNA reduced MCF10A cells. The spheroids formed by mtDNA reduced cells are dysmorphic and lose their acinar organization as observed in parental cells.
Red= Phalloidin; Blue= DAPI
Even though the cell lineage and markers of “cancer stem cells” remains contentious, it is widely accepted that in many cancers, metastatic tumors are formed by a small population of cells during an EMT, which have the tumor initiating potential. These cells are characterized and isolated by their cell surface markers which vary depending on the tumor of origin. Interestingly, mtDNA loss generates brCSC-like cells with self-renewal capacity suggesting a link between low mtDNA copy number and cellular reprogramming to “stemness” [38]. Our study made a seminal contribution in delineating a defined role for mitochondrial retrograde signaling in driving mammary tumorigenesis, from initiation to progression through EMT [38].
As described in an earlier section, EMT involves a multitude of well-coordinated and tightly regulated steps. One of the mechanisms by which the requirement of the metastasizing cell is met is by increasing the protein diversity by alternative splicing. It is therefore significant that mitochondrial retrograde signaling modulates the expression of the Epithelial Splicing Regulatory Protein (ESRP)-1, which regulates the alternative splicing of a wide array of gene sets involved in inducing EMT [38,158-166]. Another key signaling protein of mitochondrial retrograde signaling pathway, Akt kinase is induced in mtDNA depleted human mammary epithelial cells. The EMT specific transcription factor, Twist has been shown to upregulate Akt resulting in inducing cellular invasion and acquired cell motility [143]. This is crucial because Akt kinase controls EMT-specific alternative splicing events by phosphorylating splicing factors such as serine/arginine-rich splicing regulator SRSF1 [167]. Therefore we postulate that mtDNA defects induced-mitochondrial retrograde signaling drives breast tumor EMT via modulating global changes in the patterns of alternative splicing and thereby increasing proteomic diversity.
FUTURE DIRECTIONS FOR THERAPEUTIC BENEFIT
It seems plausible that dysfunctional mitochondria in tumor cells provide adaptive and survival advantage to metastasizing cells. Mitochondrial retrograde signaling is an upstream effector of EMT because it regulates the expression of nuclear genes involved in cellular reprogramming. Based on our studies and reports showing clinical correlations between mtDNA defects and poor prognostic outcome, we suggest that mtDNA copy number could be a useful prognostic marker.
Our evidence that mitochondrial stress signaling induces metastatic progression in an hnRNPA2-and ESRP1-dependent manner implies changes in alternative splicing events play a crucial role in this signaling driven EMT during breast cancer metastasis. Therefore gene isoforms that are altered specifically by low mtDNA during metastatic transition of primary tumors will suggest that therapeutics which target splicing machinery currently under clinical trials could be more beneficial to patients with primary breast tumors containing low mtDNA. In future studies, it needs to be explored whether reduction in mtDNA copy numbers in some primary tumors is due to defects in mitochondrial DNA transcription or translation and identify the drug-targetable factors to correct such defects. Reports that breast cancer cell lines lacking mtDNA (rho zero) showed decreased sensitivity to chemotherapeutic drugs such as doxorubicin, vincristine and paclitaxel [168], suggest that patients with low mtDNA content tumors are resistant to chemotherapeutic agents and accounts for the poor prognosis of breast cancer patients.
ACKNOWLEDGMENTS
This work was supported by the Breast Cancer Alliance grant 568489 (to M.G.) and NIH grant CA-22762 (to N.G.A.)
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- Christofori G (2006) New signals from the invasive front. Nature 441: 444-450.
- Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100: 57-70.
- Fidler IJ (2003) The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer 3: 453-458.
- Weiss L (2000) Metastasis of cancer: a conceptual history from antiquity to the 1990s. Cancer Metastasis Rev 19: 193-383.
- Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin Invest 119: 1420-1428.
- Kang Y, Massagué J (2004) Epithelial-mesenchymal transitions: twist in development and metastasis. Cell 118: 277-279.
- Thiery JP (2002) Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2: 442-454.
- Thiery JP, Acloque H, Huang RY, Nieto MA (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139: 871-890.
- Thiery JP, Sleeman JP (2006) Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 7: 131-142.
- Mareel M, Leroy A (2003) Clinical, cellular, and molecular aspects of cancer invasion. Physiol Rev 83: 337-376.
- Steeg PS (2006) Tumor metastasis: mechanistic insights and clinical challenges. Nat Med 12: 895-904.
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, et al. (2003) Cell migration: integrating signals from front to back. Science 302: 1704-1709.
- Webb DJ, Parsons JT, Horwitz AF (2002) Adhesion assembly, disassembly and turnover in migrating cells -- over and over and over again. Nat Cell Biol 4: 97-100.
- Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, et al. (2006) The consensus coding sequences of human breast and colorectal cancers. Science 314: 268-274.
- Azab AK, Hu J, Quang P, Azab F, Pitsillides C, et al. (2012) Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood 119: 5782-5794.
- Cannito S, Novo E, Compagnone A, Valfrè di Bonzo L, Busletta C, et al. (2008) Redox mechanisms switch on hypoxia-dependent epithelial-mesenchymal transition in cancer cells. Carcinogenesis 29: 2267-2278.
- Chen J, Imanaka N, Chen J, Griffin JD (2010) Hypoxia potentiates Notch signaling in breast cancer leading to decreased E-cadherin expression and increased cell migration and invasion. Br J Cancer 102: 351-360.
- Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, et al. (2007) Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest 117: 3810-3820.
- Jiang J, Tang YL, Liang XH (2011) EMT: a new vision of hypoxia promoting cancer progression. Cancer Biol Ther 11: 714-723.
- Copple BL (2010) Hypoxia stimulates hepatocyte epithelial to mesenchymal transition by hypoxia-inducible factor and transforming growth factor-beta-dependent mechanisms. Liver Int 30: 669-682.
- Luo D, Wang J, Li J, Post M (2011) Mouse snail is a target gene for HIF. Mol Cancer Res 9: 234-245.
- Jing Y, Han Z, Zhang S, Liu Y, Wei L (2011) Epithelial-Mesenchymal Transition in tumor microenvironment. Cell Biosci 1: 29.
- Zhou Q, Zeng R, Xu C, Liu L, Chen L, et al. (2012) Erbin inhibits TGF-β1-induced EMT in renal tubular epithelial cells through an ERK-dependent pathway. J Mol Med (Berl) 90: 563-574.
- Pettit EJ, Fay FS (1998) Cytosolic free calcium and the cytoskeleton in the control of leukocyte chemotaxis. Physiol Rev 78: 949-967.
- Komuro H, Rakic P (1992) Selective role of N-type calcium channels in neuronal migration. Science 257: 806-809.
- Lee J, Ishihara A, Oxford G, Johnson B, Jacobson K, et al. (1999) Regulation of cell movement is mediated by stretch-activated calcium channels. Nature 400: 382-386.
- Marks PW, Maxfield FR (1990) Transient increases in cytosolic free calcium appear to be required for the migration of adherent human neutrophils. J Cell Biol 110: 43-52.
- Nishiyama M, Hoshino A, Tsai L, Henley JR, Goshima Y, et al. (2003) Cyclic AMP/GMP-dependent modulation of Ca2+ channels sets the polarity of nerve growth-cone turning. Nature 423: 990-995.
- Yang S, Huang XY (2005) Ca2+ influx through L-type Ca2+ channels controls the trailing tail contraction in growth factor-induced fibroblast cell migration. J Biol Chem 280: 27130-27137.
- Lewis RS (2007) The molecular choreography of a store-operated calcium channel. Nature 446: 284-287.
- Parekh AB, Fleig A, Penner R (1997) The store-operated calcium current I (CRAC): nonlinear activation by InsP3 and dissociation from calcium release. Cell 89: 973-980.
- Yang S, Zhang JJ, Huang XY (2009) Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 15: 124-134.
- Huang S, Chong N, Lewis NE, Jia W, Xie G, Garmire LX, et al. (2016) Novel personalized pathway-based metabolomics models reveal key metabolic pathways for breast cancer diagnosis. Genome Med 8: 34.
- Cuyàs E, Corominas-Faja B, Menendez JA (2014) The nutritional phenome of EMT-induced cancer stem-like cells. Oncotarget 5: 3970-3982.
- Guha M, Fang JK, Monks R, Birnbaum MJ, Avadhani NG (2010) Activation of Akt is essential for the propagation of mitochondrial respiratory stress signaling and activation of the transcriptional coactivator heterogeneous ribonucleoprotein A2. Mol Biol Cell 21: 3578-3589.
- Guha M, Srinivasan S, Biswas G, Avadhani NG (2007) Activation of a novel calcineurin-mediated insulin-like growth factor-1 receptor pathway, altered metabolism, and tumor cell invasion in cells subjected to mitochondrial respiratory stress. J Biol Chem 282: 14536-14546.
- Srinivasan S, Guha M, Dong DW, Whelan KA, Ruthel G, et al. (2016) Disruption of cytochrome c oxidase function induces the Warburg effect and metabolic reprogramming. Oncogene 35: 1585-1595.
- Guha M, Srinivasan S, Ruthel G, Kashina AK, Carstens RP, et al. (2014) Mitochondrial retrograde signaling induces epithelial-mesenchymal transition and generates breast cancer stem cells. Oncogene 33: 5238-5250.
- Kondaveeti Y, Guttilla Reed IK, White BA (2015) Epithelial-mesenchymal transition induces similar metabolic alterations in two independent breast cancer cell lines. Cancer Lett 364: 44-58.
- Briege KJ (2006) Embryonic transcription factors in human breast cancer. IUBMB Life 58: 123-132.
- Grego-Bessa J, Díez J, Timmerman L, de la Pompa JL (2004) Notch and epithelial-mesenchyme transition in development and tumor progression: another turn of the screw. Cell Cycle 3: 718-721.
- Hiscox S, Jiang WG, Obermeier K, Taylor K, Morgan L, et al. (2006) Tamoxifen resistance in MCF7 cells promotes EMT-like behaviour and involves modulation of beta-catenin phosphorylation. Int J Cancer 118: 290-301.
- Müller T, Bain G, Wang X, Papkoff J (2002) Regulation of epithelial cell migration and tumor formation by beta-catenin signaling. Exp Cell Res 280: 119-133.
- Mani SA, Yang J, Brooks M, Schwaninger G, Zhou A, et al. (2007) Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc Natl Acad Sci USA 104: 10069-10074.
- Moody SE, Perez D, Pan TC, Sarkisian CJ, Portocarrero CP, et al. (2005) The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell 8: 197-209.
- Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, et al. (2010) Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci USA 107: 15449-15454.
- Vandewalle C, Comijn J, De Craene B, Vermassen P, Bruyneel E, et al. (2005) SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Res 33: 6566-6578.
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, et al. (2004) Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117: 927-939.
- Yook JI, Li XY, Ota I, Hu C, Kim HS, et al. (2006) A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol 8: 1398-1406.
- Bracken CP, Khew-Goodall Y, and Goodall GJ (2015) Network-Based Approaches to Understand the Roles of miR-200 and Other microRNAs in Cancer. Cancer Res. 75: 2594-2599.
- Guttilla IK, Adams BD, White BA (2012) ERα, microRNAs, and the epithelial-mesenchymal transition in breast cancer. Trends Endocrinol Metab 23: 73-82.
- Roy SS, Gonugunta VK, Bandyopadhyay A, Rao MK, Goodall GJ, et al. (2014) Significance of PELP1/HDAC2/miR-200 regulatory network in EMT and metastasis of breast cancer. Oncogene 33: 3707-3716.
- Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature 490: 61-70.
- Charafe-Jauffret E, Ginestier C, Monville F, Fekairi S, Jacquemier J, et al. (2005) How to best classify breast cancer: conventional and novel classifications (review). Int J Oncol 27: 1307-1313.
- Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, et al. (2015) Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 163: 506-519.
- Desmedt C, Haibe-Kains B, Wirapati P, Buyse M, Larsimont D, et al. (2008) Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin Cancer Res 14: 5158-5165.
- Sørlie T (2004) Molecular portraits of breast cancer: tumour subtypes as distinct disease entities. Eur J Cancer 40: 2667-2675.
- Sørlie T, Wang Y, Xiao C, Johnsen H, Naume B, et al. (2006) Distinct molecular mechanisms underlying clinically relevant subtypes of breast cancer: gene expression analyses across three different platforms. BMC Genomics 7: 127.
- Walker RA, Varley JM (1993) The molecular pathology of human breast cancer. Cancer Surv 16: 31-57.
- Wirapati P, Sotiriou C, Kunkel S, Farmer P, Pradervand S, et al. (2008) Meta-analysis of gene expression profiles in breast cancer: toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res 10: 65.
- Tian F, Wang Y, Seiler M, Hu Z (2014) Functional characterization of breast cancer using pathway profiles. BMC Med Genomics 7: 45.
- Xu T, Le TD, Liu L, Wang R, Sun B, et al. (2016) Identifying Cancer Subtypes from miRNA-TF-mRNA Regulatory Networks and Expression Data. PLoS One 11: 0152792.
- Lee H, Palm J, Grimes SM, Ji HP (2015) The Cancer Genome Atlas Clinical Explorer: a web and mobile interface for identifying clinical-genomic driver associations. Genome Med 7: 112.
- Lim E, Vaillant F, Wu D, Forrest NC, Pal B, et al. (2009) Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med 15: 907-913.
- Campbell CT, Kolesar JE, Kaufman BA (2012) “Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number”. Biochim Biophys Acta 1819: 921-929.
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, et al. (2005) Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309: 481-484.
- Warburg O (1956) On the origin of cancer cells. Science 123: 309-314.
- Seyfried TN (2015) Cancer as a mitochondrial metabolic disease. Front Cell Dev Biol 3: 43.
- Seyfried TN, Huysentruyt LC (2013) On the origin of cancer metastasis. Crit Rev Oncog 18: 43-73.
- Clayton DA, Vinograd J (1967) Circular dimer and catenate forms of mitochondrial DNA in human leukaemic leucocytes. Nature 216: 652-657.
- Clayton DA, Vinograd J (1969) Complex mitochondrial DNA in leukemic and normal human myeloid cells. Proc Natl Acad Sci USA 62: 1077-1084.
- Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, et al. (2001) Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet 69: 49-54.
- Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, et al. (2000) Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287: 848-851.
- Brière JJ, Favier J, Bénit P, El Ghouzzi V, Lorenzato A, et al. (2005) Mitochondrial succinate is instrumental for HIF1alpha nuclear translocation in SDHA-mutant fibroblasts under normoxic conditions. Hum Mol Genet 14: 3263-3269.
- Guzy RD, Sharma B, Bell E, Chandel NS, Schumacker PT (2008) Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol 28: 718-731.
- Hensen EF, Siemers MD, Jansen JC, Corssmit EP, Romijn JA, et al. (2011) Mutations in SDHD are the major determinants of the clinical characteristics of Dutch head and neck paraganglioma patients. Clin Endocrinol (Oxf) 75: 650-655.
- Janeway KA, Kim SY, Lodish M, Nosé V, Rustin P, et al. (2011) Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci USA 108: 314-318.
- Kim S, Kim do H, Jung WH, Koo JS (2013) Succinate dehydrogenase expression in breast cancer. Springerplus 2: 299.
- Niemann S, Müller U (2000) Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet 26: 268-270.
- Owens KM, Aykin-Burns N, Dayal D, Coleman MC, Domann FE, et al. (2012) Genomic instability induced by mutant Succinate Dehydrogenase Subunit D (SDHD) is mediated by O2(-•) and H2O2. Free Radic Biol Med 52: 160-166.
- Paik JY, Toon CW, Benn DE, High H, Hasovitz C, et al. (2014) Renal carcinoma associated with succinate dehydrogenase B mutation: a new and unique subtype of renal carcinoma. J Clin Oncol 32: 10-13.
- Pollard PJ, Brière JJ, Alam NA, Barwell J, Barclay E, et al. (2005) Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet 14: 2231-2239.
- Ricketts C, Woodward ER, Killick P, Morris MR, Astuti D, et al. (2008) Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst 100: 1260-1262.
- Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, et al. (2005) Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7: 77-85.
- Castro-Vega LJ, Buffet A, De Cubas AA, Cascón A, Menara M, et al. (2014) Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet 23: 2440-2446.
- Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, et al. (2005) HIF over expression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8: 143-153.
- Launonen V, Vierimaa O, Kiuru M, Isola J, Roth S, et al. (2001) Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci USA 98: 3387-3392.
- Sudarshan S, Sourbier C, Kong HS, Block K, Valera Romero VA, et al. (2009) Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1alpha stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol 29: 4080-4090.
- Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, et al. (2002) Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 30: 406-410.
- Tong WH, Sourbier C, Kovtunovych G, Jeong SY, Vira M, et al. (2011) The glycolytic shift in fumarate-hydratase-deficient kidney cancer lowers AMPK levels, increases anabolic propensities and lowers cellular iron levels. Cancer Cell 20: 315-327.
- Y Yang Y, Valera VA, Padilla-Nash HM, Sourbier C, Vocke CD, et al. (2010) UOK 262 cell line, Fumarate Hydratase deficient (FH-/FH-) hereditary leiomyomatosis renal cell carcinoma: in vitro and in vivo model of an aberrant energy metabolic pathway in human cancer. Cancer Genet Cytogenet 196: 45-55.
- Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, et al. (2011) IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol 224: 334-343.
- Borger DR, Goyal L, Yau T, Poon RT, Ancukiewicz M, Deshpande V, et al. (2014) Circulating oncometabolite 2-hydroxyglutarate is a potential surrogate biomarker in patients with isocitrate dehydrogenase-mutant intrahepatic cholangiocarcinoma. Clin Cancer Res 20: 1884-1889.
- Borger DR, Tanabe KK, Fan KC, Lopez HU, Fantin VR, et al. (2012) Frequent mutation of Isocitrate Dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 17: 72-79.
- Cairns RA, Iqbal J, Lemonnier F, Kucuk C, de Leval L, et al. (2012) IDH2 mutations are frequent in angioimmunoblastic T-cell lymphoma. Blood 119: 1901-1903.
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, et al. (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462: 739-744.
- Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, et al. (2010) Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med 207: 339-344.
- Kang MR, Kim MS, Oh JE, Kim YR, Song SY, et al. (2009) Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int J Cancer 125: 353-355.
- Kato Kaneko M, Liu X, Oki H, Ogasawara S, Nakamura T, et al. (2014) Isocitrate dehydrogenase mutation is frequently observed in giant cell tumor of bone. Cancer Sci 105: 744-748.
- Kloosterhof NK, Bralten LB, Dubbink HJ, French PJ, van den Bent MJ (2011) Isocitrate dehydrogenase-1 mutations: a fundamentally new understanding of diffuse glioma? Lancet Oncol 12: 83-91.
- Liu X, Kato Y, Kaneko MK, Sugawara M, Ogasawara S, et al. (2013) Isocitrate dehydrogenase 2 mutation is a frequent event in osteosarcoma detected by a multi-specific monoclonal antibody MsMab-1. Cancer Med 2: 803-814.
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, et al. (2009) Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 361: 1058-1066.
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, et al. (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321: 1807-1812.
- Parekh AB, Fleig A, Penner R (1997) The store-operated calcium current I (CRAC): nonlinear activation by InsP3 and dissociation from calcium release. Cell 89: 973-980.
- Shibata T, Kokubu A, Miyamoto M, Sasajima Y, Yamazaki N (2011) Mutant IDH1 confers an in vivo growth in a melanoma cell line with BRAF mutation. Am J Pathol 178: 1395-1402.
- Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, et al. (2010) The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17: 225-234.
- Watanabe T, Nobusawa S, Kleihues P, Ohgaki H (2009) IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 174: 1149-1153.
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, et al. (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360: 765-773.
- Bardella C, Pollard PJ, Tomlinson I (2011) SDH mutations in cancer. Biochim Biophys Acta 1807: 1432-1443.
- Letouzé E, Martinelli C, Loriot C, Burnichon N, Abermil N, et al. (2013) SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 23: 739-752.
- Xiao M, Yang H, Xu W, Ma S, Lin H, et al. (2012) Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 26: 1326-1338.
- Bell EL, Emerling BM, Ricoult SJ, Guarente L (2011) SirT3 suppresses hypoxia inducible factor 1α and tumor growth by inhibiting mitochondrial ROS production. Oncogene 30: 2986-2996.
- Finley LW, Carracedo A, Lee J, Souza A, Egia A, et al. (2011) SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Cancer Cell 19: 416-428.
- Huang KH, Hsu CC, Fang WL, Chi CW, Sung MT, et al. (2014) SIRT3 expression as a biomarker for better prognosis in gastric cancer. World J Surg 38: 910-917.
- Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, et al. (2010) SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 17: 41-52.
- Xiao K, Jiang J, Wang W, Cao S, Zhu L, et al. (2013) Sirt3 is a tumor suppressor in lung adenocarcinoma cells. Oncol Rep 30: 1323-1328.
- Zhang YY, Zhou LM (2012) Sirt3 inhibits hepatocellular carcinoma cell growth through reducing Mdm2-mediated p53 degradation. Biochem Biophys Res Commun 423: 26-31.
- Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ (2003) Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem 278: 36027-36031.
- Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, et al. (2008) ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 320: 661-664.
- Sharma LK, Fang H, Liu J, Vartak R, Deng J, et al. (2011) Mitochondrial respiratory complex I dysfunction promotes tumorigenesis through ROS alteration and AKT activation. Hum Mol Genet 20: 4605-4616.
- Wallace DC (1994) Mitochondrial DNA mutations in diseases of energy metabolism. J Bioenerg Biomembr 26: 241-250.
- Wallace DC (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 39: 359-407.
- Zhou G, Dada LA, Wu M, Kelly A, Trejo H, et al. (2009) Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am J Physiol Lung Cell Mol Physiol 297: 1120-1130.
- Dong R, Wang Q, He XL, Chu YK, Lu JG, et al. (2007) “Role of nuclear factor kappa B and reactive oxygen species in the tumor necrosis factor-alpha-induced epithelial-mesenchymal transition of MCF-7 cells”. Braz J Med Biol Res 40: 1071-1078.
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, et al. (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133: 704-715.
- Moreno-Bueno G, Portillo F, Cano A (2008) Transcriptional regulation of cell polarity in EMT and cancer. Oncogene 27: 6958-6969.
- Nieto MA, Cano A (2012) The epithelial-mesenchymal transition under control: Global programs to regulate epithelial plasticity. Semin Cancer Biol 22: 361-368.
- Fan AX, Radpour R, Haghighi MM, Kohler C, Xia P, et al. (2009) Mitochondrial DNA content in paired normal and cancerous breast tissue samples from patients with breast cancer. J Cancer Res Clin Oncol 135: 983-989.
- Hsu CW, Yin PH, Lee HC, Chi CW, Tseng LM (2010) Mitochondrial DNA content as a potential marker to predict response to anthracycline in breast cancer patients. Breast J 16: 264-270.
- Imanishi H, Hattori K, Wada R, Ishikawa K, Fukuda S, et al. (2011) Mitochondrial DNA mutations regulate metastasis of human breast cancer cells. PLoS One 6: 23401.
- Lee HC, Yin PH, Lin JC, Wu CC, Chen CY, et al. (2005) Mitochondrial genome instability and mtDNA depletion in human cancers. Ann N Y Acad Sci 1042: 109-122.
- Lee HC, Yin PH, Lin JC, Wu CC, Chen CY, et al. (2005) Mitochondrial genome instability and mtDNA depletion in human cancers. Ann N Y Acad Sci 1042: 109-122.
- Salgado J, Honorato B, García-Foncillas J (2008) Review: mitochondrial defects in breast cancer. Clin Med Oncol 2: 199-207.
- Tseng LM, Yin PH, Chi CW, Hsu CY, Wu CW, et al. (2006) Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. Genes Chromosomes Cancer 45: 629-638.
- Yu M, Zhou Y, Shi Y, Ning L, Yang Y, et al. (2007) Reduced mitochondrial DNA copy number is correlated with tumor progression and prognosis in Chinese breast cancer patients. IUBMB Life 59: 450-457.
- McMahon S, LaFramboise T (2014) Mutational patterns in the breast cancer mitochondrial genome, with clinical correlates. Carcinogenesis 35: 1046-1054.
- Cai FF, Kohler C, Zhang B, Chen WJ, Barekati Z, et al. (2011) Mutations of mitochondrial DNA as potential biomarkers in breast cancer. Anticancer Res 31: 4267-4271.
- Zhu W, Li WY, Kuang AK, Tan MG, Qin JF, et al. (1983) A preliminary study on serum trace elements in “Yin-deficiency” and “Yang-deficiency” patients. Application of the PIXE analysis in medical science. J Tradit Chin Med 3: 145-150.
- Canter JA, Kallianpur AR, Parl FF, Millikan RC (2005) Mitochondrial DNA G10398A polymorphism and invasive breast cancer in African-American women. Cancer Res 65: 8028-8033.
- Dimberg J, Hong TT, Nguyen LT, Skarstedt M, Löfgren S, et al. (2015) Common 4977 bp deletion and novel alterations in mitochondrial DNA in Vietnamese patients with breast cancer. Springerplus 4: 58.
- Shen L, Fang H, Chen T, He J, Zhang M, et al. (2010) Evaluating mitochondrial DNA in cancer occurrence and development. Ann N Y Acad Sci 1201: 26-33.
- Ye C, Shu XO, Pierce L, Wen W, Courtney R, et al. (2010) Mutations in the mitochondrial DNA D-loop region and breast cancer risk. Breast Cancer Res Treat 119: 431-436.
- Ye C, Shu XO, Wen W, Pierce L, Courtney R, et al. (2008) Quantitative analysis of mitochondrial DNA 4977-bp deletion in sporadic breast cancer and benign breast diseases. Breast Cancer Res Treat 108: 427-434.
- Porteous WK, James AM, Sheard PW, Porteous CM, Packer MA, et al. (1998) Bioenergetic consequences of accumulating the common 4977-bp mitochondrial DNA deletion. Eur J Biochem 257: 192-201.
- Ma Y, Bai RK, Trieu R, Wong LJ (2010) Mitochondrial dysfunction in human breast cancer cells and their transmitochondrial cybrids. Biochim Biophys Acta 1797: 29-37.
- Kaipparettu BA, Ma Y, Park JH, Lee TL, Zhang Y, et al. (2013) Crosstalk from non-cancerous mitochondria can inhibit tumor properties of metastatic cells by suppressing oncogenic pathways. PLoS One 8: 61747.
- Amuthan G, Biswas G, Ananadatheerthavarada HK, Vijayasarathy C, Shephard HM, et al. (2002) Mitochondrial stress-induced calcium signaling, phenotypic changes and invasive behavior in human lung carcinoma A549 cells. Oncogene 21: 7839-7849.
- Amuthan G, Biswas G, Zhang SY, Klein-Szanto A, Vijayasarathy C, et al. (2001) Mitochondria-to-nucleus stress signaling induces phenotypic changes, tumor progression and cell invasion. EMBO J 20: 1910-1920.
- Biswas G, Adebanjo OA, Freedman BD, Anandatheerthavarada HK, Vijayasarathy C, et al. (1999) Retrograde Ca2+ signaling in C2C12 skeletal myocytes in response to mitochondrial genetic and metabolic stress: a novel mode of inter-organelle crosstalk. EMBO J 18: 522-533.
- Biswas G, Anandatheerthavarada HK, Zaidi M, Avadhani NG (2003) Mitochondria to nucleus stress signaling: a distinctive mechanism of NFkappaB/Rel activation through calcineurin-mediated inactivation of IkappaBbeta. J Cell Biol 161: 507-519.
- Biswas G, Guha M, Avadhani NG (2005) Mitochondria-to-nucleus stress signaling in mammalian cells: nature of nuclear gene targets, transcription regulation, and induced resistance to apoptosis. Gene 354: 132-139.
- Clayton DA (1998) Nuclear-mitochondrial intergenomic communication. Biofactors 7: 203-205.
- Guha M, Avadhani NG (2013) Mitochondrial retrograde signaling at the crossroads of tumor bioenergetics, genetics and epigenetics. Mitochondrion 13: 577-591.
- Guha M, Pan H, Fang JK, Avadhani NG (2009) Heterogeneous nuclear ribonucleoprotein A2 is a common transcriptional coactivator in the nuclear transcription response to mitochondrial respiratory stress. Mol Biol Cell 20: 4107-4119.
- Guha M, Tang W, Sondheimer N, Avadhani NG (2010) Role of calcineurin, hnRNPA2 and Akt in mitochondrial respiratory stress-mediated transcription activation of nuclear gene targets. Biochim Biophys Acta 1797: 1055-1065.
- Wallace DC (2012) Mitochondria and cancer. Nat Rev Cancer 12: 685-698.
- Jiang H-L, Sun H-F, Gao S-P, Li LD, Huang S, et al. (2016) SSBP1 Suppresses TGFβ-Driven Epithelial-to-Mesenchymal Transition and Metastasis in Triple-Negative Breast Cancer by Regulating Mitochondrial Retrograde Signaling. Cancer Res 76: 952.
- Srinivasan S, Avadhani NG (2007) Hypoxia-mediated mitochondrial stress in RAW264.7 cells induces osteoclast-like TRAP-positive cells. Ann N Y Acad Sci 1117: 51-61.
- Bebee TW, Park JW, Sheridan KI, Warzecha CC, Cieply BW, et al. (2015) The splicing regulators Esrp1 and Esrp2 direct an epithelial splicing program essential for mammalian development. Elife 4.
- Cieply B, Carstens RP (2015) Functional roles of alternative splicing factors in human disease. Wiley Interdiscip Rev RNA 6: 311-326.
- Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, et al. (2011) An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet 7: 1002218.
- Warzecha CC, Carstens RP (2012) Complex changes in alternative pre-mRNA splicing play a central role in the Epithelial-to-Mesenchymal Transition (EMT). Semin Cancer Biol 22: 417-427.
- Warzecha CC, Hovhannisyan R, Carstens RP (2012) Dynamic fluorescent and luminescent reporters for cell-based splicing screens. Methods Mol Biol 867: 273-287.
- Warzecha CC, Jiang P, Amirikian K, Dittmar KA, Lu H, et al. (2012) An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition. Embo J 29: 3286-3300.
- Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP (2009) ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell 33: 591-601.
- Warzecha CC, Shen S, Xing Y, Carstens RP (2009) The epithelial splicing factors ESRP1 and ESRP2 positively and negatively regulate diverse types of alternative splicing events. RNA Biol 6: 546-562.
- Yang Y, Park JW, Bebee TW, Warzecha CC, Guo Y, et al. (2016) Determination of a comprehensive alternative splicing regulatory network and the combinatorial regulation by key factors during the epithelial to mesenchymal transition. Mol Cell Biol.
- Blaustein M, Pelisch F, Tanos T, Muñoz MJ, Wengier D, et al. (2005) Concerted regulation of nuclear and cytoplasmic activities of SR proteins by AKT. Nat Struct Mol Biol 12: 1037-1044.
- Yu M, Shi Y, Wei X, Yang Y, Zang F, et al. (2009) Mitochondrial DNA depletion promotes impaired oxidative status and adaptive resistance to apoptosis in T47D breast cancer cells. Eur J Cancer Prev 18: 445-457.
Citation: Guha M, Avadhani NA (2016) Epithelial To Mesenchymal Transition in Breast Cancer Metastasis: Mitochondria Take the Center Stage. J Cell Biol Cell Metab 3: 009.
Copyright: © 2016 Manti Guha, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.