Essential Impact of Antibodies and Drug Conjugates in Cancer Therapy
*Corresponding Author(s):
Muhammad KalimDepartment Of Microbiology & Immunology, Wake Forest School Of Medicine, Winston-Salem, NC 27101, United States
Tel:336-995-6999,
Email:mkalim@wakehealth.edu
Abstract
Cancer is one of the leading causes of death worldwide. Cancer cells exhibit several characteristics, including high growth rates, reduced apoptosis, and abnormal metabolic activity[1]. Therapeutic ability play a fundamental role in the treatment of cancer. Antibodies often show limited effectiveness and must be bound to a functional molecule, drug, or agent to significantly improve efficacy. Antibody-drug conjugates (ADCs) have the advantage of monoclonal antibodies thar deliver drugs specifically and precisely to target tumor cells. Considerable achievements have been fulfilled by using the conjugation strategy of ADC. Each ADC component (mAb, linker, and drug) should simply be considered fully functional to accomplish its goal of improving therapeutic effect and reducing toxicity. Existing challenges include improving therapeutic index, choosing appropriate target, elucidating immune responses, ADC trafficking, next-generation molecular advances, and considerate clinical environment to bring the highest clinical and sub-clinical achievements. Subsequent efforts were concisely reviewed in detail to assess the functional role of ADCs and its conjugates in therapeutic developments.
Keywords
Antibody; Endocytosis; Antibody-drug conjugates; Cancer therapy; Immunity; Bio-pharmaceutics
Abbreviations
ADC: Antibody-Drug Conjugate
FDA: Food and Drug Administration
CD: Cluster of Differentiation
MMAE: Monomethyl Auristatin
ADCC: Antibody-Dependent Cellular Cytotoxicity
CDC: Complement Dependent Cytotoxicity
EGFR: Epidermal Growth Factor Receptor
VEGF: Vascular Endothelial Growth Factor
sALCL: Systematic Anaplastic Large Cell Lymphoma
CTLA: Cytotoxic T Lymphocyte-Associated proteins
APC: Antigen Presenting Cell
DC: Dendritic Cell
CLL: Chronic Lymphocytic Leukemia
FcRn: Neonatal Fc Receptor
ITIMs: Immune Receptor Tyrosine-based Inhibitory Motifs
LDL: Low-Density Lipoprotein
DAR: Drug-Antibody Ratio
ILV: Intra-luminal Vesicle
Introduction
Cancer causes the most considerable mortality rate worldwide. Cancerous cells show numerous characteristics of increased proliferation rate, reduce apoptosis, and abnormal metabolic activities[1]. To control the malignant transformation of these cells, a lot of efforts have been made to examine the genomic studies, metabolites, signaling pathways, and mechanistic activity of drugs related to cancer cells [1]. Historically, the first description of the cancer-related disease was recorded in Egyptian papyri during 1600 BC. Advanced radiation therapy and surgery accelerated in the first half of the twentieth century, but most were inadequate to fight metastatic cancer. In 1940, nitrogen mustard was considered anti-neoplastic chemotherapy that target all tumor cells [2]. The concept of selective targeting of tumor cells with antibody conjugate or targeting agents comes from the German Physician Paul Ehrlich, who coined the term ‘magic bullet’ [3]. The concept of selective targeting was first epitomized 50 years later when methotrexate was linked to an antibody that target leukemia cells. Kohler and Landmark then significantly improved the ADC history to develop mouse mAbs using hybridoma technology [4]. In addition, human mAbs became available in the 1990s using genetically modified mice and phage display technology [5]. Antibody-drug conjugates (ADC) are a rapidly expanding class of anti-cancer therapeutics, consisting of an antibody attached, via a chemical linker, to a potent cytotoxic drug also called payload. The antibody is designed precisely to target a specific antigen that is highly expressed in tumor cells. The majority of ADCs follows the same mode of action that involves antibody mediated receptor attachment, internalization, and release of cytotoxic payloads.
The U.S. Food and Drug Administration approved the first ADC, the anti-mitotic vinca alkaloid vindesine (Mylotarg, gemtuzumab ozogamicin), in clinical trials in 2000, but discontinued in 2010 due to its limited efficacy [6]. As of July 2021, the U.S. Food and Drug Administration has approved 11 ADCs and more than 80 ADCs are currently under active clinical development that can selectively eliminate the tumor cells through specific or associated antigens [7-10]. Majority of these drugs utilized auristatin and maytansinoids antimitotic agents [11]. Comprehensive studies of molecular mechanisms, intracellular trafficking, designing of efficient agents of ADC, and their interaction with tumor cells must be needed for the development of efficient ADCs. The current work covers recent advances of ADCs in clinical therapeutics and its counterparts. As illustrated, linker strategies and binding affinity became a key role in the successful optimization of ADC.
Antibody Drug Conjugates
ADCs are vital bio-pharmaceutics designed for cancer therapy and intended to targets the cancer cells leaving the normal cells. Four antibody-drug conjugates have been approved by US FDA so far as shown in Table 1. Mylotarg (gemtuzumab ozogamicin) was approved for the treatment of AML in 2000 [12]. This drug comprises an anti-CD33 antibody conjugated with DNA strand break calicheamicin. Mylotarg was withdrawn from the market in June 2010 but reintroduced in markets of the US in 2017 [13,14]. Brentuximab vedotin (Adcetris) and ado-trastuzumab emtansine (T-DM1, Kadcyla) were the next approved drugs for the treatment of cancers. Adectris make conjugation of anti-CD30 antibody with monomethyl auristatin (MMAE), were approved in 2011 for the treatment of Hodgkin’s lymphoma [15]. Kadcyla, an anti-HER3 antibody conjugated with maytansinoid DM1, was approved in 2013 for the treatment of metastatic breast cancers [16]. The newly approved ADC, Inotuzumab ozogamicin, is calicheamicin conjugated with anti-CD22 mAb that results in a double-strand break in CD22 positive acute lymphoblastic leukemia (ALL) [7]. ADCs have three main parts: a monoclonal antibody, a linker, and a cytotoxic drug. All these components greatly affect the performance rate of ADC and its optimization (Figure 1) [17]. Additionally, the developmental progress of ADC depends extensively on better knowledge of antibody components of ADC and its trafficking in the intracellular region of targeted tumor cells [18]. Recently, some studies indicated that an engineered antibody can be utilized to exploit the endosomal pathways that provide a substantial clove for future studies and better designing of ADC. The experimental analysis provides knowledge of the intracellular process in greater aspects dissolves recent divergences and enhances the ability to select novel and efficient targets for ADCs attachment. Additional vital research analysis must be needed to parallel analysis, like studies of tumor cells toxicity, target receptors modification studies, and cascade signaling analysis of receptors modulation by antibodies.
More comprehensive knowledge must be provided by this information and additionally deliver a broad understanding of proper balance maintenance between target antigens and its sympathy for ADC.
|
ADC Names |
Antibody |
Antigen |
Linker/cytotoxic agent |
Tumor |
Developer |
Status |
|
Zynlonta |
Humanized IgG1 |
CD19 |
Valanie-alanine/ SG3249 PDB dimer |
B-cell lymphoma |
ADC Therapeutics |
Approved 2021 |
|
Blenrep |
Humanized IgG1 |
BCMA |
Maleimidocaproyl/ IMMAF |
Multiple myeloma |
GSK |
Approved 2020 |
|
Trodelvy |
Humanized IgG1 |
Trop-2 |
Hydrolysable CL2A/SN-38 Topo I inhibitor |
Metastatic triple-negative breast cancer |
Immunomedics |
Approved 2020 |
|
Enhertu |
Humanized IgG1 |
Her2 |
Tetrapeptide/exatecan derivatives topoisomerase I inhibitor (DXd) |
HER2-positive breast cancer |
Daiichi Sankyo |
Approved 2019 |
|
Packev |
Human IgG1 |
Nectin-4 |
Valine citrulline/MMAE |
Adult patient with locally advanced or metastatic urothelial cancer |
Astellas Pharma, Inc. |
Approved 2019 |
|
Polivy |
Humanized (IgG4) |
CD79b |
Valine citrulline/MMAE |
Relapsed/refractor diffuse B-cell lymphoma (DBCL) |
Genetech |
Approved 2019 |
|
Besponsa |
Humanized (IgG4) |
CD22 |
AcuBut/Calicheamicin |
Acute Lymphoblastic Leukemia (ALL) |
Pfizer |
Approved 2017 |
|
Inotuzumab ozogamicin (CMC-544; Besponsa) |
Inotuzumab (Humanized) |
CD22 |
Cleavable Hydrazone, AcBu N-acetyl-gamma-calicheamicin |
Acute Lymphoblastic Leukemia (ALL) |
Pfizer |
Approved by US FDA in 8/2017 |
|
|
|
|
(ozogamicin) |
|
|
|
|
Brentuximab vedotin Adcetris; SGN35)
|
Brentuximab (Chimeric IgG1) |
CD30 |
Dipeptide cleavable, vc-MMAE (auristatin) |
Relapsed/refractory Hodgkin lymphoma and systemic anaplastic large cell lymphoma |
Millennium-Takeda Seattle Genetics |
Approved by US FDA in 8/2011. Phase 1-4 multiple studies. Monotherapy and combination in CD30 positive tumors |
|
Gemtuzumab ozogamicin (Mylotarg) |
Gemtuzumab (Humanized IgG4) |
CD33 |
Hydrazone, AcBu, N-acetyl-c calicheamicin |
Acute myeloid leukemia (AML) |
Pfizer |
Approved by US FDA approval in 5/2000. Withdrawn 8/2011, Reintroduced in 2017 |
|
Ado-trastuzumab emtansine (Kadcyle; T-DM1) |
Trastuzumab (Humanized IgG1) |
HER2; ErbB2 |
Thioether (Non-cleavable) SMCC-DM1 (maytansinoid) |
Human Epidermal Growth Recptor-2 positive (HER-2) Breast Cancer |
Immuno Gen ADC technology (Genentech-Roche) |
Approved in 2/1/2013 by US FDA. Multiple Phase 1–4 studies Combination and monotherapy |
|
Inotuzumab ozogamicin (CMC-544) |
Inotuzumab (Humanized IgG4) |
CD22 |
Hydrazone cleavable, AcBu N-acetyl-c calicheamicin |
B-ALL and other B cell malignancies |
Pfizer |
In Phase 3 monotherapy and combination trials |
|
Rovalpituzumab tesirine (Rova-T; SC16LD6.5) |
Rovalpituzumab (Humanized IgG1 |
DLL3 (Delta-like protein 3) |
valine-alanine (cleavable dipeptide) PDB dimer |
SCLC |
Stemcentrx (Spirogen) |
Phase 2 trials in combination for SCLC late studies |
|
Glembatumumab vedotin (CDX-011; CR011- vcMMAE) |
Glembatumumab (Huamn IgG1) |
Glycoprotein (osteoactivin) |
vc-MMAE (cleavable dipeptide) auristatin |
MBC (Metastatic breast cancer) and melanoma |
Seattle Genetics CelldexTherapeutics |
Phase 2 in combination in triple negative breast cancer |
|
Anetumab ravtansine (BAY 94-9343) |
Anetumab (Human IgG1) |
Mesothelin |
Cleavable disulfide SPDB- DM4 (maytansinoid) |
Mesothelin-positive solid tumors |
Bayer Healthcare (ImmunoGen) |
Phase 2 mono- and combination therapy in mesothelin positive |
|
Depatuxizumab mafodotin (ABT-414) |
Depatuxizumab Humanized IgG1 |
EGRF |
Non-cleavable, maleimido- caproyl mcMMAF (auristatin) |
Solid tumors, glioblastoma expressing EGFR |
Abbvie (Seattle Genetics) |
Phase 1-3 trials in combination with temozolomide ongoing |
|
Vadastuximab talirine (SGN-CD33A) |
Vadastuximab (Humanized IgG1) |
CD33 |
valine-alanine (cleavable disulfide) PDB dimer |
AML |
Seattle Genetics (Spirogen) |
Phase 3 trials in combination with azacitidine or decitabine in AML patients |
|
Mirvetuximab soravtansine (IMGN853) |
Mirvetuximab Humanized IgG1 |
Folate receptor (Fra; FOLR1) |
Highly charged cleavable disulfide, Sulfo-SPDB-DM4 (maytansinoid) |
Lung adeno, endometrial and ovarian cancer |
ImmunoGen |
Phase 2 & 3 trial in combination platinum-resistant ovarian (FRa-positive) cancer |
Table 1: List of selected antibody drug conjugates showing its counter parts.
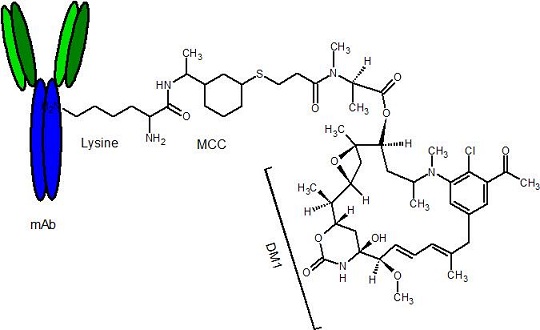 Figure 1: Representative structure of an ADC presenting monoclonal antibody, linker, and drug. The amino acid group lysine presents the active specific binding.
Figure 1: Representative structure of an ADC presenting monoclonal antibody, linker, and drug. The amino acid group lysine presents the active specific binding.
Antibody engineering and its specificity
The ADC molecules are theoretically more complex than either a monoclonal antibody or its constituents Therapeutic agents are designed to target the tumor cells specifically with the help of monoclonal antibodies. Specific antigens localize at the surface of tumor cells make complex, get internalization through endocytosis and incorporate into endosomes. Complex degradation process and enzymatic reactions take place inside the cancer cells, break down the ADC complex, and releases the toxic agents. Antibody enhances the delivery of effective drugs to targeted tumor cells. ADC is being guided by an antibody to target the tumor cell. The cell surface protein is recognized by an antibody that provides an address for the therapeutic agent. The most frequently used monoclonal antibody isotype is IgG1 [19].
The antibody-based cancer treatment is intensively studied and well appreciated that it has direct or indirect effects, on cancer cells. The restriction of tumor cells occurs through signal inhibition, limiting proliferation, apoptosis induction, cytotoxic drugs or radiation delivery, induction and activation of immune cells, and cytotoxicity, cell inhibition of payload delivered to the targeted area [20]. Therapeutic agents are designed in such a way that target the tumor cells specifically with the help of monoclonal antibodies [21,22]. These antibodies enhance the delivery of effective drugs to targeted tumor cells. The mAbs have been applied to treat various cancerous organs like the breast, kidney, colorectal, lungs, head, neck, and circulations [23]. Various studies have been reported that trastuzumab (Herceptin) is humanized IgG1 that can bind to HER2 and improve the survival rate of breast cancer patients by inhibiting protein signaling by promoting the antibody-dependent cellular cytotoxicity (ADCC). Three mAbs have been reported to target colorectal cancer antigen EGFR or VEGF [24]. Colorectal cancer was recorded the third most common in the world. Similarly, three different unconjugated mAbs are being approved by FDA to treat hematological malignancies. Rituaumab (chimeric IgG1) targets CD20 on the surface B cell of non-Hodgkin lymphoma and chronic lymphocytic leukemia that trigger lysis through apoptosis, ADCC, and CDC [24-26]. Obinutuzumab and Ofatumumab (human IgG1) specifically target the extracellular loops of the same CD20 to treat CLL patients [27]. Gemtuzumab is humanized IgG4 and Brentuximab is chimeric IgG1, are drug conjugates that target CD33 to treat the hematological malignancies and relapsed or refractory Hodgkin lymphoma and sALCL (systematic anaplastic large cell lymphoma) [28,29].
Four different mechanisms of inhibitions were reported by antibodies against tumor cells. These are a perturbation of tumor cell signaling, activation of CDC (complement-dependent cytotoxicity), ADCC (antibody-dependent cellular cytotoxicity), and adaptive immunity induction (Figure 2) [30].
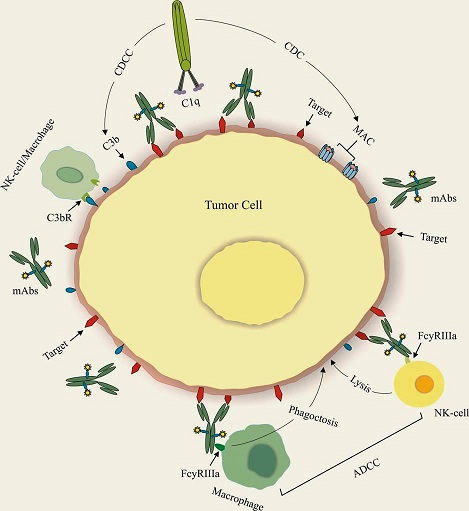 Figure 2: Mechanism of antibody action in tumor cell disruption using CDC, CDC and ADCC signaling.
Figure 2: Mechanism of antibody action in tumor cell disruption using CDC, CDC and ADCC signaling.
Perturbation of signaling
Efficient antibodies were developed to target cytokines. From these regions, they can start their antagonistic activities. Growth factor receptors such as EGFR (Epidermal Growth Factor Receptor) are expressed on tumor cells which are inhibited by antibodies to stop their mitogenic signaling [31]. Similarly, CTLA-4 was reported to suppress immune receptors or increase antigenic presentation on APCs by activating receptors as CD40 [32,33].
Complement-dependent cytotoxicity (CDC)
Complement comprised of more than 30 proteins, assembled to kill a foreign cell through MAC by proteolytic cascade complex. Thus, it activates inflammatory response via anaphylatoxin and eliminates targets [34,35]. Two or more antibodies get attach to the cell and activate the classical complement pathway via the C1 complex to antibody Fc domains. It activates cascade mechanisms of phagocytosis and lysis [36]. CDC significantly contributes to the activity of therapeutic antibodies against tumor cells. Rituximab targets CD20 positive cells, is a potent activator of CDC, and use to treat many B cell malignancies [37]. Ofatumumab is type I anti-CD20 antibody, attaches specifically to stimulate CDC more than rituximab [38]. It was reported that ofatumumab has a greater affinity to attach with C1q than rituximab showed enhanced efficiency B cell lymphoma cell lines that resist tuximab [39]. Clinically ofatumumab showed enhanced response against refractory chronic lymphocytic leukemia (CLL), thus get approval in 2009 by FDA [27].
Antibody-dependent cell-mediated cytotoxicity (ADCC)
Activation of ADCC by antibody Fc region via FcRs of immune cells was also reported. Immune receptor tyrosine-based activation motifs (ITAMs) and immune receptor tyrosine-based inhibitory motifs (ITIMs) stimulated via FcRs. Three types of FcRs, FcRI (CD64), FcRIIA (CD32A), and FcRIIIA (CD16A) exits in mediated cytotoxicity. Single inhibitory receptor, FcRIIB (CD32B) and FcRIIIA were reported in natural killer cells to persist prominent effector cells of ADCC [40]. However, mediation of granulocyte cells and macrophages has been recorded to a lesser extent. Antibody-coated target cells can be recognized by these infected cells via FcRs, release perforin and granzymes results in cell lysis [30].
Adaptive immunity induction
A number of researchers recommended that adaptive immunity induction showed importance to the antibody. Antibody helps to generate adaptive immunity via CDC and ADCC [41]. These generate tumor cell fragment, release antigens and taken by APC such as DC, to initiate tumor-directed adaptive immunity. It also generates triggering Fc dependent phagocytosis using opsonin [42]. DCs processed the tumor antigens through endocytosis, presents MHC II to prime CD4+ T cells. Additionally, CDs also help to generate CD8+ cytotoxic T cells by cross-presentation. Cytotoxic T cell kills tumor cells directly or further differentiate into tumor-specific T memory cell [43].
Antibody Implications in ADC Development
Characteristics of the monoclonal antibody are critical attribute to ensure the final ADC complex recognition. The ideal surface antigen must ensure the active transport of loaded drugs to the internal lumen of the cell. Physical and chemical properties of mAbs have been extensively studied and found its steadiness compared to other proteins [44,45]. Selection of well-characterized antigens greatly increases the opportunity to develop a successful ADC. Also, the full expression pattern throughout the healthy and tumor cells must be considered evading the toxic side effects. Several tumor-associated antigens have been developed as prospective targets for the antibody component of ADCs. Leukemia, lymphoma, and multiple myeloma cancer types help in the study of ADCs. B cell and T cell surface proteins are most preferred to target, as they are extensively expressed on the surface of B and T cells in these types [46].
Ackerman et al. reported that ADC efficacy can be affected by receptor endocytosis. They studied two antibodies M85151a and M111147 which can bind to CEA (carcinoembryonic antigen). As M85151a recognizes two epitopes and thus cross-links carcinoembryonic antigen, lead to more sharply internalization [47]. When these antibodies were incubated with LS174T spheroids, it came to noticed that the slower M111147 penetrate more rapidly than the M85151a antibody. Thus developing ADC, analysis of receptor internalization mechanics, its effects and delivery of ADC were prominently noticed [48].
Two recent works were employed to enhance the serum clearance half-life by using mutagenic strategy. Different pH binding affinity was used. In the first study, TCZ (tocilizumab) was modified to target the interleukin 6 receptor (IL-6R). TCZ is promoted to treat moderate to severe RA (Rheumatoid Arthritis) [49]. The interleukin-6 receptor goes through a high rate of membrane turnover cause cellular clearance of TCZ via lysosomal degradation and an efficient decrease in plasma removal half-life. So researchers of this study limit the TCZ binding affinity to interleukin-6 receptors of endosomal pH 6.0 without its effect on pH 7.4 at the membrane surface. Thus, it allowed tocilizumab to unlink from degradation in lysosomes so interleukin gets in, attach FcRn and recycle back on membrane surface [50]. Histidine scanning approach is being utilized by the investigator to re-engineer the binding affinity of tocilizumab at pH 6.0 which involve mutation of critical amino acid. PCSK9 (proprotein convertase subtilisin kexin type 9) antibody is also re-engineered by other investigators to enhance the degradation of the LDL receptor which directly increases the LDL- cholesterol [51].
Recycling of receptors such as transferrin, LDL, integrin, metabotropic glutamate 5, and HER2 takes place to membrane surfaces after its cellular internalization [52-54]. By developing ADC against these receptors, one possible attempt to engineer the prominent antibodies that demonstrate the reasonable returned to surfaces. While, for some ligands such as tumor growth factor α it is not applicable as they can certainly dissociate from their receptor in the endosome, traffic to the lysosome and degrade there. This method has no need of neonatal Fc receptor, as it is present in the fluid phase of endosomes and can transport IgG out from inside lumen to membrane surface [55-57].
The Linker Binding Strategies
Linkers play pivotal roles in ADC design to maintain the balance and efficiency in the bloodstream. Initially, linkers like hydrazone were designed in such a way that releases the toxic compartment in the acidic environment only. However, limited target actions, instability in circulation, failure to detach, multiple striking rounds and triggering side effects were reported in these types of linkers. Linker helps ADCs to conjugate a cytotoxic drug with a monoclonal antibody and should be stable in circulation to allow the efficient release of carrying the drug, once ADC gets into tumor cell [58]. Two types of the linker are being utilized to develop ADC. These are cleavable (hydrazine or disulfide) and non-cleavable thioether) (Figure 3). Cleavable linkers release its carrying drugs through the splitting of the bond between them [8,59] while in non-cleavable linker all elements attached with each other, followed by proteolytic degradation in lysosome [60-62].
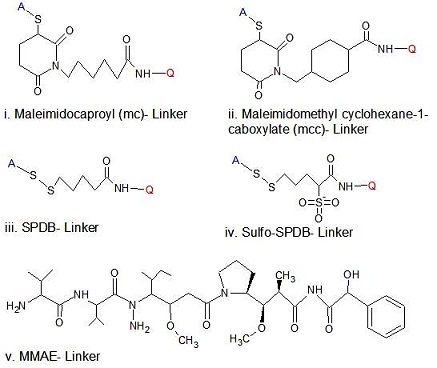 Figure 3: Generic structure of linkers that are used in marketed approved ADCs.
Figure 3: Generic structure of linkers that are used in marketed approved ADCs.
The first ADC, Gemtuzumab ozogamicin (Mylotarg), was constructed using hydrazone linker with calicheamicin. Currently, two other candidates, inotuzumab ozogamicin (CMC-544) and milatuzumab doxorubicin as shown in Fig. 4 utilize hydrazone linker for the treatment of lymphoid malignancies [63]. The majority of ADC utilizes direct attachment of linker with a cysteine residue of the antibody by taking advantage of maleimide activity with sulfhydryl groups. One disadvantage of maleimide chemistry for ADC is chemical instability in circulation that results in the premature release of drugs. It was reported that thiosuccinimide hydrolysis can prevent the premature release, by incorporation of the amino group to maleimide [64]. Heterogenous development of ADCs was generated by incorporation of amino acid groups (lysine or cysteine) with antibody residues (Figure 1). The approach of amino acid binding leads to efficient suboptimal properties and optimization of ADC challenges [65]. The initially available linkage chemistry of linker to a polypeptide of antibodies was restricted to the natural amino acid side chain, as NH2 in lysine and SH in cysteine. Lysine conjugation completed in two steps of first binding with antibody and then with drugs. Roughly 90 lysine residues were found in the IgG1 antibody and 30 residues modified by the different condition for experimental and clinical purposes (http://www.lifetechnologies.com/us/en/home/references/molecular-probes-the-handbook.html). Formation of heterogeneous mixture occurred by lysine-binding and characterized by drug antibody ratio (DAR), typically 3-4 residues [66]. These drugs can be attached to any of 30 available lysine residues. Cysteine binding developed disulfide-bridge to create Cys-SH under controlled condition. As there are 8 possible conjugation sites on a typical IgG1 antibody, creating a lower heterogeneity compared to lysine conjugation. These interactions of lysine and cysteine with antibodies still possess a challenge for ADC optimization and its potency for cancer treatment. Recently phage display technology developed thiols residue THIOMAB, which do not alter binding specificity or function [67].
The Cytotoxic Agent Potency In ADC Efficacy
Calicheamicin, duocarmycins, maytansinoids, anthracycline, and auristatins are high evaluated drugs used as cytotoxic components of ADCs. Calicheamicin and duocarmycins cause irreversible DNA damage while others result in sorting out of cell microtubule structural fibers. Auristatin derivatives (MMAE & MMAF) and maytansine derivatives (DM1 & DM4) result in the disruption of microtubules and eventually cell death [68]. These agents possess high potency and efficiency in targeted therapy. If even less dose gets into tumor cells, it will liberate maximum effect as reported in maytansine derivatives. These are 100 to 10,000 more efficient than a standard chemotherapeutic agent [69]. Various drugs can bind to single monoclonal antibody and its drug antibody (DAR) ratios affect the pharmacokinetic studies and distribution astonishingly [70]. Most cytotoxic compounds were found hydrophobic in nature and linking them with antibodies, creates aggregation problems and thus limit drug loading leading to liver toxicity and elevated immunogenicity [71,72]. Additionally, the hydrophobic nature of most compounds creates multidrug resistance response and exit the cytotoxic compound out of cell compromised the efficacy of ADC. However, it was reported that using a hydrophobic linker with maytansinoids produced DM1 hydrophilic metabolites with no resistance and markedly effective ADC [73].
Doxorubicin comprised from anthracycline family was first hemi-synthesized compound isolated from Streptomyces in 1960 and were markedly approved in 1974 for the treatment of several cancers. It was conjugated with a hydrazone acid labile linker and cBR96 monoclonal antibody to minimize its toxicity. This conjugate releases its drug due to the elevated acidic environment of endosome and lysosome as compared to circulation. Preclinical study of doxorubicin conjugated ADC showed limited clinical profile and safety but showed elevated efficacy as compared to naked antibody [74].
DNA alkylating agent, Calicheamicin, was isolated in 1986 from Micromonospora, presents enedilyne moiety and allylic trisulfide group that results in DNA cleavage [75]. Gentuzumab was first approved ADC utilized calicheamicin drug moiety to treat relapsed acute myeloid leukemia in 2000 [12,76]. The conjugate presents two to three DAR via hydrazone linker and releases its drug by hydrolysis. Pre-mature release in circulations was restrained by addition of N-acetyl-calicheamicin dimethyl hydrazide to the antibody via an acid-labile linker. Inotuzumab ozogamicin (CMC-544) also utilized calicheamicin derivative to link with anti-CD22 monoclonal antibody [77]. This ADC molecule utilizes hydrazine linker appears more stable in circulation. Additional optimization studies are evaluating in CMC-544 to treat acute lymphoblastic leukemia patients [78].
Auristatins are the most prominent linkers utilized in ADC development. Both synthetic MMAE and MMAF of dolastatin series were originally discovered as a wedge sea hare Dolabella auricularia and modified to auristatins suitable for ADC development and conjugation [79]. These drugs prevent the polymerization of mitotic apparatus. Brentuximab vedotin (SGN-33) utilized auristatin for clinical evaluation, approved markedly in 2011 (Figure 4) [79,80]. Another new dolastatin derivative, tubulysins, was reported a potent mitotic inhibitor that was derived from myxobacteria Archangium gephyra and Angiococcus disciformis [81]. Additionally, ADCs comprise emtansine and maytansine conjugates, accounts emerging cytotoxic drugs that cover 80% of current drug discovery platform. Amanita mushroom was reported for the production of α-amantin that inhibits DNA and RNA translocation by blocking RNA polymerase II activities [82]. Amantin sites were found suitable for linker integration and ADC optimization, ADC-DSC- α-amantin conjugate, in ERB2, overexpressing tumor cell lines. A single dose can remit tumor inclusion in SKOV-3 and JIMT-1 human xenograft model [83,84]. The intrinsic safety measures and liver response for α-amantin are still unclear.
 Figure 4: Structure of selected approved ADCs and its counterparts.
Figure 4: Structure of selected approved ADCs and its counterparts.
Internalization Mechanism And Conjugation Strategies Of ADC
ADCs are designed in such a way that retains stability in circulation and maintained constancy of antibody and its loaded drugs. Linker stability can largely impact its payload release mechanism. ADC specially binds with tumor surface antigen via its antibody, internalized into cancer cells and subjected to hydrolysis by low pH environment, disulfide reduction or proteases to release its carrying drugs [18,85]. Lysosomal Cathepsin B has been reported to involve in several drug release by its proteolytic enzymatic activity. The lysosomal low pH (4.6-5.0) also results in hydrolysis for various linkers as in hydrazone linkers. ADC internalization initiated with binding to surface antigens or ligands on tumor cells. These targeted antigens may include carbohydrates, small surface ligands, antibodies, proteins, and peptides. Different pathways facilitate the internalization of ADCs. These pathways include the prominent clathrin pathways, additionally less prominent caveolae-mediated and cholesterol-dependent macropinocytosis pathways [18]. The carrying cargo gets fused with early endosomes where ADCs sorted to various pathways of continuously dropping pH from 6.2-6.3 in early endosome to 4.6-5.0 in lysosomes (Figure 5). The enzymatic activity of lysosomes and pH environments helps in the endocytic escape of carrying drugs that largely depends on linker chemistry [86].
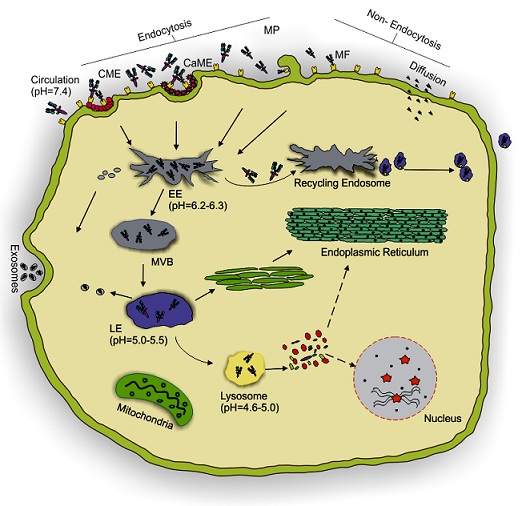 Figure 5: Mechanism of endocytosis and intracellular trafficking. The mechanism includes (a) endocytosis and (b) non-endocytosis. The ADCs from circulations (pH 7.4) are mediated by clathrin and caveolae to early endosomes (EE) where pH drops to 6.2-6.3. Non-endocytosis involves direct membrane fusion (MF) or diffusion via the surface membrane. ADCs either recycle back to the membrane from EE via recycling endosomes to surface or directed to the multi-vesicular body (MVB). MVB either mature to late endosome (LE) (pH 5.0-5.5) or as exosomes. LE fuses with the lysosome (pH 4.6-5.0) to lyse the ADCs into its components and release its active contents for action. CME: Clathrin Mediated Endocytosis, CaME: Caveolae Mediated Endocytosis, MP: Macropinocytosis.
Figure 5: Mechanism of endocytosis and intracellular trafficking. The mechanism includes (a) endocytosis and (b) non-endocytosis. The ADCs from circulations (pH 7.4) are mediated by clathrin and caveolae to early endosomes (EE) where pH drops to 6.2-6.3. Non-endocytosis involves direct membrane fusion (MF) or diffusion via the surface membrane. ADCs either recycle back to the membrane from EE via recycling endosomes to surface or directed to the multi-vesicular body (MVB). MVB either mature to late endosome (LE) (pH 5.0-5.5) or as exosomes. LE fuses with the lysosome (pH 4.6-5.0) to lyse the ADCs into its components and release its active contents for action. CME: Clathrin Mediated Endocytosis, CaME: Caveolae Mediated Endocytosis, MP: Macropinocytosis.
The essential chemical and biological concerns of heterogeneous mixtures have directed to achieve site-specific conjugation that enables precise binding of loaded drugs with antibodies. These achievements can be avail by engineered cysteine residues, non-natural amino acids incorporation, and enzymatic modification [66]. Standard cysteine residues are utilized to developed engineered cysteine, while non-natural amino acids have been developed by enzymatic alteration of full monoclonal antibody [87,88].
Endosomal Escape and Drug Release
In the cytoplasm, endosomal vesicles fuse with each other to form early endosome once they detached from the membrane. This early endosome gives warm welcome to incoming ADC molecules also with fluid from membrane plasma [89]. Its internal environment pH ranges from 5.9 to 6.8 and provides initial categorization compartment for internalized ADC [90-92]. The endocytosed ADC can be recycled back by short recycling loop to the cell membrane or can be propelled to the Golgi complex for repackaging by using early endosome retromer complex [93-95]. Endosomal maturation takes place from early endosome to late endosomes which are also called MVBs (multi-vesicular bodies) [96]. This maturation is characterized by a rise in luminal acidification and formation of ILVs (intra-luminal vesicles). ILVs contain the specific ADC with a potent drug which was budded up from late endosome [97,98]. This ADC complex delivered to the lumen of lysosome where the potent drug is released and ready to show its potency and targeting the specific sites (Figure 5). The discovery of trafficking processes greatly facilitates the efficacy of engineered antibody drug conjugates [21,22].
Efficient release of potent drugs in the cytosol of the cell is another vital factor that affects the efficiency of ADC. The release of the drug is due to breakage of a chemical bond between antibody and drug, the linker. Thus linker choices have a great impact on ADC designing. Hydrazone linkers are acid labile, stable at neutral pH of extracellular environment and cleaved intracellular endosome and lysosome environment of low pH [99]. Hydrolysis of hydrazone linkers results in liberation of hydrazine and aldehyde [100]. These types of linker release the loaded drug calicheamicin as in gemtuzuman ozogamicin. Sulfide anion formation followed by thiophene ring development takes place by degradation of calicheamicin disulfide bond that results in alkylation of DNA and cell lysis [101]. Inotuzumab ozogamicin and milatuzumab doxorubicin are two new ADCs under investigation, utilize hydrazone linker but no studies found related to their drug release strategies and toxicity response [102,103].
Citrulline valine is a dipeptide linker which is selectively broken by lysosomal proteases including cathepsin B [104]. The dipeptide linkers were reported more stable compared to others [105]. Brentuxumab vedotin comprised of MMAE conjugated with CD30 mAb via dipeptide linker. This ADC releases MMAE elimination of p-aminobenzyl carbamate by cathepsin B-mediated dipeptide breakage [86]. The premature release of the val-cit linker was reported in plasma membrane due to unknown serine proteases [106]. Engineered cysteine was developed to link the SGN-CD33A antibody to pyrrolobenzodiazepine (PDB)- DNA binding agent and showed systemic stability in circulation [86]. The cytotoxic payload is liberated upon internalization in target expressing cells.
Similarly, non-cleavable linkers are also being developed which are stable throughout the plasma membrane and intracellular space. Its payload liberation relies on, degradation in lysosomes following antigen-mediated internalization as in thioether linkers [107]. T-DM1 (Kadcyla, trastuzumab emtansine) utilizes non-cleavable linker is one of US FDA approved ADC. It was reported that proteolytic cleavage of T-DM1 release liberation of MCC-DM1 and lysine-SMCC-DM1 two catabolites [62,108]. Maleimidocaproyl linker is another example of the non-cleavable linker that connects the anti-CD30 monoclonal antibody with anti-mitotic auristatin, MMAF [109]. Intracellular degradation of this linker results catabolite containing lysine or cysteine residues. Careful selection is required as all catabolites are not active, however, MMAF conjugates with non-cleavable linker fulfill the potency of activity [86].
Self-immolating disulfide linkers were developed recently that make conjugation directly with CD22 antibody through cysteine thiol residues. This novel drug release mechanism reduces disulfide and self-immolates to thiol intermediate followed by the release of drug and other catabolites [110]. Cyclobutyl and methyl analogs of thiol reduction resulted in PDB production and cyclopropyl analog reduction results in inactive thiol catabolites. The cellular toxicity was reported less than 50-fold for cyclopropyl as compared to other two also the pKa value (4.8) was significantly less than cyclobutyl analog (pKa= 9.6) and this may be due to non-immolation results less efficacy [111].
Next generation ADCs
Efficient engineering of ADC can evaluate the efficacy and limit the toxicity of the toxic dose as compared to tradition chemotherapy. This targeted therapy of cytotoxic agent to cancer cells increases the delivery to cells thus minimized the effective dose and increasing the tolerated dose. The off targeted toxicity of second generation development also narrowed the therapeutic windows. The reported toxicities of active drugs, as well as optimization of antibodies, linkers, and drugs, have driven the rational approach of third generation ADC development.
First generation drug conjugates (Gemtuzumab ozogamicin, Mylotarg) showed unsatisfactory results and was withdrawn in mid-2010. The drug calicheamicin was conjugated with the anti-CD33 monoclonal antibody. Failures of Mylotarg were no improvement in survival and higher toxicity rate. Some studies were also reported by targeting both CD33 positive and negative cells that result in targets effectiveness by targeting CD33+ cells. Recently, it was warmly welcomed to the complexity of these molecules and to develop increasingly sophisticated and pure ADCs to treat tumor cells. The second generation anti-CD33 ADC, AVE9633, were developed by conjugation of DM4 with a humanized IgG1 anti CD33 monoclonal antibody against AML. In AVE9633, multidrug resistance-associated proteins 1 (MDR1) does not seem to be involved in resistance as were reported previously in Gemtuzumab ozogamicin. Discontinuation of this drug occurs due to its poor activity. The low density of antibody CD33 also results in insufficient delivery of DM1 to interrupt G2/M alteration in AML. The third generation ADC vadastuximab talirine (SGN-CD33A) was developed that causes crosslinking of DNA and blocking cell division. SGN-CD33A contains synthetic PDB dimer conjugated with humanized anti-CD33. Site-specific conjugation occurs due to the addition of cysteine residues on heavy chains that also deliver targeted drugs concisely and mimic toxicity [112]. The second and third generation agents show a great ability to treat cancer cells. All second-generation species are a mixture of loaded drugs of specific ratio with antibodies as 3.5 for trastuzumab emantansine and 4 for brentuximab ozogamicin. Most of ADCs in clinical trials share the same structural characteristics. Both cysteine and lysine-linked ADC possess greater toxicity and off targeted release of drugs in rapid physiological conditions [64,113]. These unusual conditions can be resolved by alteration binding chemistries and site-specific conjugations as the development of engineered cysteines, engineering of unnatural amino acids, assisted ligation of enzymes, redesigning of glycan and glyco-conjugation, serine terminal modification and highly loaded ADCs with more than 8 DAR [114].
Conclusion
ADCs are designed in such a way to mimic the degradation in circulation and successfully deliver its potent loaded drugs to targeted areas. Degradation of ADC in an intracellular environment has a great impact on efficacy and pharmacokinetics. Catabolism of ADC may be altered by linker types, drug-antibody ratios, and conjugation strategies. Maleimide linkers mostly dependent on cellular internal environment and solvent availability while disulfide linkers tend to conjugate with plasma proteins. Linker diversification and warhead improvement have delivered novel prospects to improve precise and potent drug delivery to targeted cells. Consideration of these factors will improve stable ADC designing and efficient access of ADCs to target intact. Significant research efforts are now being directed towards site-specific conjugation strategies of ADCs. These strategies rely either on engineered cysteine residues or non-amino acid conjugated with additional functional groups such as azide, ketone, alkynyl or aldehyde. The site-specific conjugation of antibody alteration also increases the homogeneity of ADC that lead to potent linker development and efficient ADC generation.
However further investigation is needed to optimize ADC components, improve its efficacy and toxicity, evaluate conjugation chemistry and enhance quantitative translocation systems improvements. Alternative formats of monoclonal antibodies are needed to discover the efficacy and potency of ADC. These included single chain fragments, protein peptides, dual conjugates, Pro bodies and Fabs [115]. Use of engineered conjugation sites for the better control, stability and enhanced therapeutic effects of magic bullets also provide a window for investigation of a naval agent. ADC development will seek to optimize the designing and improve effects to provide more safety and potent entity.
References
- Hayes DF (2013) OMICS-based personalized oncology: If it is worth doing, it is worth doing well! Bmc Medicine 11: 221.
- Papac RJ (2001) Origins of cancer therapy. Yale Journal of Biology and Medicine 74: 391-398.
- Bechhold H, Ehrlich P (1906) Connections between chemical constitution and desinfection effect. Article on the study of "inner antisepsis". Hoppe-Seylers Zeitschrift Fur Physiologische Chemie 47: 173-199.
- Kohler G, Milstein C (1975) Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256: 495-497.
- Lonberg N (2005) Human antibodies from transgenic animals. Nature Biotechnology 23: 1117-1125.
- Linenberger ML, Hong T, Flowers D, Sievers EL, Gooley TA, et al. (2001) Multidrug-resistance phenotype and clinical responses to gemtuzumab ozogamicin. Blood 98: 988-994.
- Lamb YN (2017) Inotuzumab Ozogamicin: First Global Approval. Drugs 77: 1603-1610.
- Lambert JM, Morris CQ (2017) Antibody-Drug Conjugates (ADCs) for Personalized Treatment of Solid Tumors: A Review. Advances in Therapy 34: 1015-1035.
- Criscitiello CS, Morganti S, Curigliano G (2021) Antibody–drug conjugates in solid tumors: A look into novel targets. Journal of Hematology & Oncology 14: 20.
- Dean AQ, Luo S, Twomey JD, Zhang B (2021) Targeting cancer with antibody-drug conjugates: Promises and challenges. MAbs 13: 1951427.
- Beck A, Reichert JM (2014) Antibody-drug conjugates Present and future. Mabs 6: 15-17.
- Bross PF, Beitz J, Chen G, Chne XH, Duffy E, et al. (2001) Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clinical Cancer Research 7: 1490-1496.
- Petersdorf SH, Kopecky KJ, Slovak M, Willman C, Nevill T, et al. (2013) A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 121: 4854-4860.
- Godwin CD, Gale RP, Walter RB (2017) Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia 31: 1855-1868.
- Senter PD, Sievers EL (2012) The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nature Biotechnology 30: 631-637.
- Lambert JM, Chari RVJ (2014) Ado-trastuzumab Emtansine (T-DM1): An Antibody-Drug Conjugate (ADC) for HER2-Positive Breast Cancer. Journal of Medicinal Chemistry 57: 6949-6964.
- Sievers EL, Senter PD (2013) Antibody-Drug Conjugates in Cancer Therapy. Annual Review of Medicine 64: 15-29.
- Kalim M, Chen J, Wang S, Lin C, Ullah S, et al. (2017) Intracellular trafficking of new anticancer therapeutics: Antibody-drug conjugates. Drug Design Development and Therapy 11: 2265-2276.
- Deslandes A (2014) Comparative clinical pharmacokinetics of antibody-drug conjugates in first-in-human Phase 1 studies. Mabs 6: 859-870.
- Scott AM, Allison JP, Wolchok JD (2012) Monoclonal antibodies in cancer therapy. Cancer Immunity 12: 14.
- Beck A, Haeuw JF, Wurch H, Goetsch L, Bailly C, et al. (2010) The Next Generation of Antibody-drug Conjugates Comes of Age. Discovery Medicine 53: 329-339.
- Adams GP, Weiner LW (2005) Monoclonal antibody therapy of cancer. Nature Biotechnology 23: 1147-1157.
- Kinch MS (2014) An analysis of FDA-approved drugs for oncology. Drug Discovery Today 19: 1831-1835.
- Cho WCS, Roukos DH (2013) Trastuzumab emtansine for advanced HER2-positive breast cancer and beyond: Genome landscape-based targets. Expert Review of Anticancer Therapy 13: 5-8.
- Baselga J, Swain SM (2010) CLEOPATRA: A Phase III Evaluation of Pertuzumab and Trastuzumab for HER2-Positive Metastatic Breast Cancer. Clinical Breast Cancer 10: 489-491.
- Vucic EA, Thu KL, Robison K, Rybaczyk LA, Chari R, et al. (2012) Translating cancer 'omics' to improved outcomes. Genome Research 22: 188-195.
- Coiffier B, Lepretre S, Pedersen LM, Gadeberg O, Fredriksen H, et al. (2008) Safety and efficacy of ofatumumab, a fully human monoclonal anti-CD20 antibody, in patients with relapsed or refractory B-cell chronic lymphocytic leukemia: A phase 1-2 study. Blood 111: 1094-1100.
- Bross PF, Beitz J, Chen G, Chen XH, Duffy E, et al. (2001) Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clinical Cancer Research 7: 1490-1496.
- Vaklavas C, Forero-Torres A (2012) Safety and efficacy of brentuximab vedotin in patients with Hodgkin lymphoma or systemic anaplastic large cell lymphoma. Therapeutic Advances in Hematology 3: 209-225.
- Kubota T, Niwa R, Satoh M, Akinaga S, Shitara K, et al. (2009) Engineered therapeutic antibodies with improved effector functions. Cancer Science 100: 1566-1572.
- Frampton JE (2010) Cetuximab: A Review of its Use in Squamous Cell Carcinoma of the Head and Neck. Drugs 70: 1987-2010.
- French RR, Chan HT, Tutt AL, Glennie MJ (1999) CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nature Medicine 5: 548-553.
- O'Day SJ, Hamid O, Urba WJ (2007) Targeting cytotoxic T-lymphocyte antigen-4 (CTLA-4)-A novel strategy for the treatment of melanoma and other malignancies. Cancer 110: 2614-2627.
- Dunkelberger JR, Song WC (2010) Complement and its role in innate and adaptive immune responses. Cell Research 20: 34-50.
- Zipfel PF, Skerka C (2009) Complement regulators and inhibitory proteins. Nature Reviews Immunology 9: 729-740.
- Stoermer KA, Morrison TE (2011) Complement and viral pathogenesis. Virology 411: 362-373.
- Winiarska M, Glodkowska-Mrowka E, Bil J, Golab J (2011) Molecular mechanisms of the antitumor effects of anti-CD20 antibodies. Frontiers in Bioscience-Landmark 16: 277-306.
- Teeling JL, Mackus WJM, Wiegman LJJM, Brakel JHN, Beers SA, et al. (2006) The biological activity of human CD20 monoclonal antibodies is linked to unique epitopes on CD20. Journal of Immunology 177: 362-371.
- Cheson BD (2010) Ofatumumab, a Novel Anti-CD20 Monoclonal Antibody for the Treatment of B-Cell Malignancies. Journal of Clinical Oncology 28: 3525-3530.
- Ferris RL, Jaffee EM, Ferrone S (2010) Tumor Antigen-Targeted, Monoclonal Antibody-Based Immunotherapy: Clinical Response, Cellular Immunity, and Immunoescape. Journal of Clinical Oncology 28: 4390-4399.
- Hainsworth JD, Litchy S, Burries HA, Scullin DC, Corso SW, et al. (2002) Rituximab as first-line and maintenance therapy for patients with indolent non-Hodgkin's lymphoma. Journal of Clinical Oncology 20: 4261-4267.
- Albert ML, Sauter B, Bhardwaj N (1998) Dendritic cells acquire antigen from apoptotic cells and induce class I restricted CTLs. Nature 392: 86-89.
- Dhodapkar KM, Krasovsky J, Williamson B, Dhodapkar MV (2002) Antitumor monoclonal antibodies enhance cross-presentation of cellular antigens and the generation of myeloma-specific kill T cells dentritic cells. Journal of Experimental Medicine 195: 125-133.
- Manning MC, Chou DK, Murphy BM, Payne RW, Katayama DS (2010) Stability of Protein Pharmaceuticals: An Update. Pharmaceutical Research 27: 544-575.
- Chaudhuri R, Cheng Y, Middaugh CR, Volkin DB (2014) High-Throughput Biophysical Analysis of Protein Therapeutics to Examine Interrelationships between Aggregate Formation and Conformational Stability. Aaps Journal 16: 48-64.
- Beck A, Haeuw JF, Wurch T, Goetsch L, Bailly C, et al. (2010) The next generation of antibody-drug conjugates comes of age. Discovery Medicine 10: 329-339.
- Ackerman ME, Pawlowski D, Wittrup KD (2008) Effect of antigen turnover rate and expression level on antibody penetration into tumor spheroids. Molecular Cancer Therapeutics 7: 2233-2240.
- Thurber GM, Schmidt MM, Wittrup KD (2008) Antibody tumor penetration: Transport opposed by systemic and antigen-mediated clearance. Advanced Drug Delivery Reviews 60: 1421-1434.
- Mircic M, Kavanaugh A (2009) The clinical efficacy of tocilizumab in rheumatoid arthritis. Drugs of Today 45: 189-197.
- Igawa T, Ishii S, Tachibana T, Maeda A, Higuchi Y, et al. (2010) Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nature Biotechnology 28: 1203-1207.
- Chaparro-Riggers J, Laing H, Devay RM, Bai L, Sutton JE, et al. (2012) Increasing Serum Half-life and Extending Cholesterol Lowering in Vivo by Engineering Antibody with pH-sensitive Binding to PCSK9. Journal of Biological Chemistry 287: 11090-11097.
- Trivedi RR, Bhattacharyya S (2012) Constitutive internalization and recycling of metabotropic glutamate receptor 5 (mGluR5). Biochemical and Biophysical Research Communications 427: 185-190.
- Pellinen T, Ivaska J (2006) Integrin traffic. Journal of Cell Science 119: 3723-3731.
- Harari D, Yarden Y (2000) Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene 19: 6102-6114.
- Roepstorff K, Grandal MV, Henriksen L, Knudsen SLP, Lerdrup M, et al. (2009) Differential Effects of EGFR Ligands on Endocytic Sorting of the Receptor. Traffic 10: 1115-1127.
- Jones SM, Foreman SK, Shank BB, Kurten RC (2002) EGF receptor downregulation depends on a trafficking motif in the distal tyrosine kinase domain. American Journal of Physiology-Cell Physiology 282: C420-C433.
- Rocca A, Lamaze C, Subtil A, Dautry-Varsat A (2001) Involvement of the ubiquitin/proteasome system in sorting of the interleukin 2 receptor beta chain to late endocytic compartments. Molecular Biology of the Cell 12: 1293-1301.
- Chari RVJ, Miller ML, Widdison WC (2014) Antibody- Drug Conjugates: An Emerging Concept in Cancer Therapy. Angewandte Chemie-International Edition 53: 3796-3827.
- Singh R, Setiady YY, Ponte J, Kovyun YK, Lai KC, et al. (2016) A New Triglycyl Peptide Linker for Antibody-Drug Conjugates (ADCs) with Improved Targeted Killing of Cancer Cells. Molecular Cancer Therapeutics 15: 1311-1320.
- Erickson HK, Park PU, Widdison WC, Kovtun YK, Garret LM, et al. (2006) Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Research 66: 4426-4433.
- Erickson HK, Widdison WC, Mayo MF, Whiteman K, Audette C, et al. (2010) Tumor Delivery and In Vivo Processing of Disulfide-Linked and Thioether-Linked Antibody-Maytansinoid Conjugates. Bioconjugate Chemistry 21: 84-92.
- Erickson HK, Lambert JM (2012) ADME of Antibody-Maytansinoid Conjugates. Aaps Journal 14: 799-805.
- Robak T, Robak E (2014) Current Phase II antibody-drug conjugates for the treatment of lymphoid malignancies. Expert Opinion on Investigational Drugs 23: 911-924.
- Lyon RP, Setter JC, Bovee TD, Doronina SO, Hunter JH, et al. (2014) Self-hydrolyzing maleimides improve the stability and pharmacological properties of antibody-drug conjugates. Nature Biotechnology 32: 1059-1062.
- Axup JY, Bajjuri KM, Ritland M, Hutchins BM, Kim CH, et al. (2012) Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proceedings of the National Academy of Sciences of the United States of America 109: 16101-16106.
- Panowski S, Bhakta S, Raab H, Polakis P, Junutula JR (2014) Site-specific antibody drug conjugates for cancer therapy. Mabs 6: 34-45.
- Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, et al. (2008) Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nature Biotechnology 26: 925-932.
- Adair JR, Howard PW, Hartley JA, Williams DG, Chester KA (2012) Antibody-drug conjugates - a perfect synergy. Expert Opinion on Biological Therapy 12: 1191-1206.
- Bernardes GJL, Casi G, Trussel S, Hartmaan I, Schwager K, et al. (2012) A Traceless Vascular-Targeting Antibody-Drug Conjugate for Cancer Therapy. Angewandte Chemie-International Edition 51: 941-944.
- Lyon RP, Bovee TD, Doronina SO, Burke PJ, Hunter JH, et al. (2015) Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nature Biotechnology 33: 733-735.
- Jarvis LM (2012) Rethinking Antibody-Drug Conjugates. Chemical & Engineering News 90: 12.
- Joubert MK, Hokom M, Eakin C, Zhou L, Deshpande M, et al. (2012) Highly Aggregated Antibody Therapeutics Can Enhance the in Vitro Innate and Late-stage T-cell Immune Responses. Journal of Biological Chemistry 287: 25266-25279.
- Kovtun YV, Audette CA, Mayo MF, Jones GE, Doherty H, et al. (2010) Antibody-Maytansinoid Conjugates Designed to Bypass Multidrug Resistance. Cancer Research 70: 2528-2537.
- Tolcher AW, Sugarman S, Gelmon KA, Cohen R, Saleh M, et al. (1999) Randomized phase II study of BR96-doxorubicin conjugate in patients with metastatic breast cancer. Journal of Clinical Oncology 17: 478-484.
- Lee MD, Lechevalier MP, Lechevalier HA, Korshalla J, Kuck N, et al. (1987) Calicheamicins, a novel family of antitumor antibiotics: taxonomy, fermentation and biological properties. Journal of the American Chemical Society 109: 3464-3466.
- Ho RJY, Chien J (2014) Trends in Translational Medicine and Drug Targeting and Delivery: New Insights on an Old Concept-Targeted Drug Delivery with Antibody-Drug Conjugates for Cancers. Journal of Pharmaceutical Sciences 103: 71-77.
- https://pubs.rsc.org/en/content/chapter/bk9781849736015-00145/978-1-84973-601-5
- DiJoseph JF, Dougher MM, Kalyandrug LB, Armellino DC, Boghaert ER, et al. (2006) Antitumor efficacy of a combination of CMC-544 (inotuzumab ozogamicin), a CD22-targeted cytotoxic immunoconjugate of calicheamicin, and rituximab against non-Hodgkin's B-cell lymphoma. Clinical Cancer Research 12: 242-249.
- Pettit GR, Srirangam JK, Barkoczy R, Williams MD, Boyd MR, et al. (1998) Antineoplastic agents 365. Dolastatin 10 SAR probes. Anti-Cancer Drug Design 13: 243-277.
- Katz J, Janik JE, Younes A (2011) Brentuximab Vedotin (SGN-35). Clinical Cancer Research 17: 6428-6436.
- Sasse F, Steinmetz H, Heil J, Höfle G, Reichenbach H (2000) Tubulysins, new cytostatic peptides from myxobacteria acting on microtubuli - Production, isolation, physico-chemical and biological properties. Journal of Antibiotics 53: 879-885.
- Gong XQ, Nedialkov YA, Burton ZF (2004) Alpha-amanitin blocks translocation by human RNA polymerase II. Journal of Biological Chemistry 279: 27422-27427.
- Anderl J, Muller C, Heckl-Östreicher B, Wehr R (2011) Highly potent antibody-amanitin conjugates cause tumor-selective apoptosis. Cancer Research 2011: 71.
- https://patents.justia.com/patent/9233173
- Tumey LN, Rago B, Han X (2015) In vivo biotransformations of antibody-drug conjugates. Bioanalysis 7: 1649-1664.
- Jain N, Smith SW, Ghone S, Tomczuk B (2015) Current ADC Linker Chemistry. Pharmaceutical Research 32: 3526-3540.
- Junutula JR, Flagella KM, Graham RA, Parsons KL, Ha E, et al. (2010) Engineered Thio-Trastuzumab-DM1 Conjugate with an Improved Therapeutic Index to Target Human Epidermal Growth Factor Receptor 2-Positive Breast Cancer. Clinical Cancer Research 16: 4769-4778.
- Hofer T, Skeffington LR, Chapman CM, Rader C (2009) Molecularly Defined Antibody Conjugation through a Selenocysteine Interface. Biochemistry 48: 12047-12057.
- Helenius A, Marsh M (1982) Endocytosis of enveloped animal viruses. Ciba Foundation Symposium 1982: 59-76.
- van Weering JRT, Verkade P, Cullen PJ (2012) SNX-BAR-Mediated Endosome Tubulation is Co-ordinated with Endosome Maturation. Traffic 13: 94-107.
- Yamashiro DJ, Maxfield FR (1987) Acidification of morphologically distinct endosomes in mutant and wild- type Chinese hamster ovary cells. Journal of Cell Biology 105: 2723-2733.
- Maxfield FR, McGraw TE (2004) Endocytic recycling. Nature Reviews Molecular Cell Biology 5: 121-132.
- Choudhury A, Sharma DK, Marks DL, Pagano RE (2004) Elevated endosomal cholesterol levels in Niemann-Pick cells inhibit Rab4 and perturb membrane recycling. Molecular Biology of the Cell 15: 4500-4511.
- Naslavsky NR, Weigert R, Donaldson JG (2004) Characterization of a nonclathrin endocytic pathway: Membrane cargo and lipid requirements. Molecular Biology of the Cell 15: 3542-3552.
- Naslavsky NR, Weigert R, Donaldson JG (2003) Convergence of non-clathrin-and clathrin-derived endosomes involves Arf6 inactivation and changes in phosphoinositides. Molecular Biology of the Cell 14: 417-431.
- Huotari J, Helenius A (2011) Endosome maturation. Embo Journal 30: 3481-3500.
- Roederer MR, Bowser R, Murphy RF (1987) Kinetics and temperature dependence of exposure of endocytosed material to proteolytic enzymes and low pH: Evidence for a maturation model for the formation of lysosomes. Journal of Cellular Physiology 131: 200-209.
- Russell MRG, Nickerson D, Odorizzi PG (2006) Molecular mechanisms of late endosome morphology, identity and sorting. Current Opinion in Cell Biology 18: 422-428.
- Polson AG, Fenaux JC, Chan P, Chang W, Christtensen E, et al. (2019) Antibody-Drug Conjugates for the Treatment of Non-Hodgkin's Lymphoma: Target and Linker-Drug Selection. Cancer Research 69: 2358-2364.
- Kalia J, Raines RT (2008) Hydrolytic stability of hydrazones and oximes. Angewandte Chemie-International Edition 47: 7523-7526.
- Bouchard HC, Viskov C, Garcia-Echeverria C (2014) Antibody-drug conjugates-A new wave of cancer drugs. Bioorganic & Medicinal Chemistry Letters 24: 5357-5363.
- Advani A, Coiffier B, Czuczman MS, Dreyling M (2010) Safety, Pharmacokinetics, and Preliminary Clinical Activity of Inotuzumab Ozogamicin, a Novel Immunoconjugate for the Treatment of B-Cell Non-Hodgkin's Lymphoma: Results of a Phase I Study. Journal of Clinical Oncology 28: 2085-2093.
- Govindan SV, Cardillo TM, Sharkey RM, Tat F, Gold DV, et al. (2013) Milatuzumab-SN-38 Conjugates for the Treatment of CD74 Cancers. Molecular Cancer Therapeutics 12: 968-978.
- Doronina SO, Toki BE, Torgiv MY, Mendelsohn BA, Cerveny CG, et al. (2003) Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nature Biotechnology 21: 778-784.
- McCombs JR, Owen SC (2015) Antibody Drug Conjugates: Design and Selection of Linker, Payload and Conjugation Chemistry. Aaps Journal 17: 339-351.
- Dorywalska M, Strop P, Melton-Witt JA, Hasa-Morano A, Farias SE, et al. (2015) Effect of Attachment Site on Stability of Cleavable Antibody Drug Conjugates. Bioconjugate Chemistry 26: 650-659.
- Kovtun YV, Audette CA, Ye Y, Xie H, Ruberti MF, et al. (2006) Antibody-drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Research 66: 3214-3221.
- Rock BM, Tometsko ME, Patel SK, Hamblett KJ, Fanslow WC, et al. (2015) Intracellular Catabolism of an Antibody Drug Conjugate with a Noncleavable Linker. Drug Metabolism and Disposition 43: 1341-1344.
- Doronina SO, Mendelsohn BA, Bovee TD, Cerveny CG, Alley SC, et al. (2006) Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: Effects of linker technology on efficacy and toxicity. Bioconjugate Chemistry 17: 114-124.
- Pillow TH, Sadowsky JD, Zhang D, Yu SF, Rosario GD, et al. (2017) Decoupling stability and release in disulfide bonds with antibody-small molecule conjugates. Chemical Science 8: 366-370.
- Zhang D, Pillow TH, Ma Y, Cruz-Chuh JD, Kozak KR, et al. (2016) Linker Immolation Determines Cell Killing Activity of Disulfide-Linked Pyrrolobenzodiazepine Antibody-Drug Conjugates. Acs Medicinal Chemistry Letters 7: 988-993.
- Sutherland MSK, Walter RB, Jeffrey SC, Burke PJ, Yu C, et al. (2012) SGN-CD33A: A novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood 122: 1455-1463.
- Dere R, Yi JH, Lei C, Saad OM, Huang C, et al. (2013) PK assays for antibody-drug conjugates: Case study with ado-trastuzumab emtansine. Bioanalysis 5: 1025-1040.
- Beck A, Goetsch L, Dumontet C, Corvaia N (2017) Strategies and challenges for the next generation of antibody drug conjugates. Nature Reviews Drug Discovery 16: 315-337.
- Puthenveetil S, Musto S, Loganzo F, Tumey LT, Donnell CJ, et al. (2016) Development of Solid-Phase Site-Specific Conjugation and Its Application toward Generation of Dual Labeled Antibody and Fab Drug Conjugates. Bioconjugate Chemistry 27: 1030-1039.
Citation: Kalim M, Lu Y (2021) Essential Impact of Antibodies and Drug Conjugates in Cancer Therapy. J Clin Immunol Immunother 7: 068.
Copyright: © 2021 Muhammad Kalim, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.