Fibrinolysis with tPA alone is only one Third as Effective as it Should be
*Corresponding Author(s):
Victor GurewichVascular Research Laboratory, Mount Auburn Hospital, Cambridge, MA, United States
Tel:+1 6179662738,
Email:vgurewich@tsillc.net
THERAPEUTIC FIBRINOLYSIS WITH TPA
Therapeutic fibrinolysis has used tissue Plasminogen Activator (tPA) alone since 1987,when tPA was first approved for the treatment of Acute Myocardial Infarction (AMI). The use of Tpa was based on the belief that it was responsible for fibrinolysis. However, this assumption should have been put into question from the outset when tPA was found to have the same efficacy as Streptokinase (SK). This was unexpected, since SK has an indirect, less efficient mechanism of action and SK has no fibrin clot affinity, in contrast to tPA. Nevertheless, the 30-day AMI mortality with tPA and SK were identical in the first two trials.
A total of 95,740 patients were tested and only in one out of four groups in the last of the three comparative trials was a significant mortality difference found with tPA over SK found. When these findings were examined by Bayesian analysis, it was concluded that a significant difference between tPA and SK had not been established [1]. Despite these findings, tPA received FDA approval for the treatment of AMI after the third trial, and nine years later received approval for the treatment of ischemic stroke as well. It remains the only activator available.
These first trials were a forerunner to the subsequent clinical experience with tPA, which has never lived up to early expectations. Curiously, the reason for this has not been identified, as far as I know. Instead, tPA has remained the only plasminogen activator available. A triumph of commercial interests over scientific and clinical evidence.
tPA looked like it was an obvious choice for fibrinolysis because it is the only activator with a high affinity for fibrin. It binds to a specific site on the D-domain of fibrin which is close to a plasminogen binding site(see Figure). As result, a ternary complex is formed between tPA, plasminogen, and fibrin and this complex promotes plasminogen activation by tPA about 1,000-fold and initiates fibrinolysis. Therefore, this reaction requires no more than a 5 mg bolus dose of tPA, which is sufficient to activate one out of the three fibrin-bound plasminogens responsible for fully effective fibrinolysis. This highly effective reaction is limited to this one plasminogen.
Since tPA is a weak plasminogen activator in the absence of this ternary complex promotion, and has no other fibrin binding site, this step completes tPA’s function in fibrinolysis. It cannot activate the other two fibrin-bound plasminogens, which are located on the fibrin E-domain (Figure 1). As a result of it being a weak activator, when tPA is used alonehigh doses must be infused to activate the two other plasminogens. As much as 150 mg was tried but this caused intracranial hemorrhage so that 100 mg is now the standard. However, even at these doses tPA remains a weak plasminogen activator and was neversufficiently effective. It is also subject to hemorrhagic side-effects, especially when used for ischemic stroke (7% intracranial hemorrhage complications).
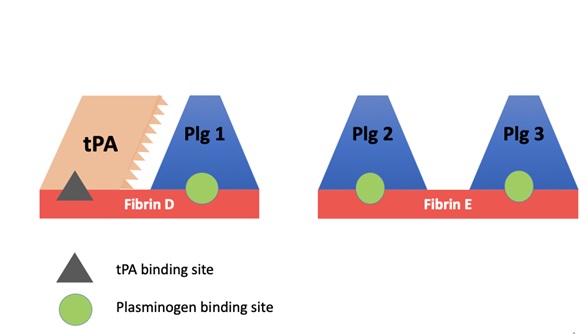 Figure 1: The plasminogen on the fibrin D-domain is activated by tPA. The two plasminogens on the E-domain are active by uPA. Plg 2 by prouPA, and Plg 3 by tcuPA (urokinase).
Figure 1: The plasminogen on the fibrin D-domain is activated by tPA. The two plasminogens on the E-domain are active by uPA. Plg 2 by prouPA, and Plg 3 by tcuPA (urokinase).
Instead, Percutaneous Coronary Intervention (PCI) has become the treatment of choice in AMI, and procedures like thrombectomy are being increasingly employed for ischemic stroke instead of or in addition to tPA. These are time-consuming hospital procedures that allow ischemic tissue damage to become irreversible. The only rapid reperfusion method available is fibrinolysis. However, both its efficacy and safety must be improved over what it is with tPA.
WHAT CAN BE LEARNED FROM ENDOGENOUS FIBRINOLYSIS? THE NATURAL DESIGN
An explanation for the inadequacy of tPA therapy is revealed by the biological design responsible for fibrinolysis. In contrast to therapy, in nature there is a second plasminogen activator in blood called urokinase Plasminogen Activator (uPA). The native form of uPA is a proenzyme, prouPA, which is stable in plasma and has fibrin-specific fibrinolytic properties [2], though it does not bind to fibrin. In physiology, tPA initiates fibrinolysis but it is then continued and completed by uPA, first by prouPA and then by two-chain uPA (tcuPA), also called urokinase (UK).
Therefore, tPA and uPA together dissolve the fibrin clot, a mechanism that is both more effective and much safer then when tPA is used alone, since the high doses Tpa that are required are associated with hemorrhagic complications duetPA binding and disrupting hemostatic fibrin sites. These are not detectable. Therefore, bleeding complications by tPA are unpredictable. They are also unnecessary, since due to tPA’s high affinity for the clot, a bolus is sufficient. Avoiding infusions essentially eliminates the bleeding risk from the lysis of hemostatic sites.
Since uPA has two active forms, prouPA and tcuPA, whereas tPA has only one, uPA is responsible for 2/3 of fibrinolysis. It activates two of the three fibrin-bound plasminogens that are responsible for fibrinolysis. The combination of bolus tPAfollowed by a prouPA infusion is also much safer. It is not only because tPA infusions are eliminated but also because significantly lower doses are required. This is because tPA and prouPA have complementary modes of action, which gives their combination a synergistic effect [3].
Nature’s combination regimen was once tested in a clinical study of 101 patients with AMI who were given a 5mg bolus of tPA followed by a 90-minute infusion of prouPA (40 mg/h) (PATENT study). Compared with the best of the tPA monotherapy studies (GUSTO), this combination regimen reduced mortality 6-fold (1% vs 6%) and almost doubled the opening of the infarct artery (82% vs 45%) [4]. These findings were consistent with the in vitro clot lysis studies showing that the combination effect to be synergistic and more effective.
Had the PATENT regimen been adopted at the time it was published, almost one million AMI deaths might have been averted in the US alone. Unfortunately, no second study with the regimen was possible since following the PATENT trial, the company developing prouPA was bought by Pharmacia which decided to discontinue its cardiovascular product line.
CONCLUSION
The use of tPA alone for fibrinolysis has been inadequate and is due to a misunderstanding of fibrinolysis. This started in 1987 when tPA was first given FDA approval. Its inadequacy is related to tPA’s fibrin binding site restricting it to the activation of only one of the three fibrin-bound plasminogens responsible for fibrinolysis. In addition, tPA monotherapy requires administration by an intravenous infusion of 100 mg, which risks causing hemorrhagic complications due to tPA binding and disrupting hemostatic fibrin. These are a vascular repair sites that are ubiquitous.
Instead, when the natural tPA paradigm is followed, no more than a 5m bolus of tPA is needed that is far safer, and which is then followed by an infusion of prouPA (a proenzyme), a combination whose efficacy is promoted by synergy. Using tPA alone has been a mistake that has been hard to reform. Although in AMI it has been replaced by PCI,this method is too time-consuming to salvage function of ischemic myocardial or brain tissue in most cases. Fibrinolysis remains the simplest and fastest method to restore circulation and restore normal function.
REFERENCES
- Brophy JM, Joseph L (1995) Placing trials in context using Bayesian analysis: GUSTO revisited by Reverend Bayes. JAMA 273: 871-875.
- Gurewich V, Pannell R, Louie S, Kelley P, Greenlee R (1984) Effective and fibrin-specific clot lysis by a zymogen precursor of urokinase (pro-urokinase) in vitro and in two animal species. J Clin Invest 73: 1731-1779.
- Pannell R, Black J, Gurewich V (1988) The complementary modes of action of tissue plasminogen activator (tPA) and pro-urokinase (proUK) by which their synergistic effect in clot lysis can be explained. J Clin Invest 81: 853-859.
- Zarich SW, KowalchukGJ, Weaver WD, Loscalzo J, Sassower M, et al. (1995) Sequential combination thrombolytic therapy for acute myocardial infarction: results of the pro-urokinase and tPA enhancement of the thrombolysis (PATENT). J Am Coll Cardiol 26:374-379.
Citation: Gurewich V (2020) Fibrinolysis with tPA alone is only one Third as Effective as it Should be. J Angiol Vasc Surg 5: 049.
Copyright: © 2020 Victor Gurewich, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.