First Case of Bilateral Fibular Aplasia, Tibial Campomelia and Oligodactyly Syndrome (FATCO Syndrome)
*Corresponding Author(s):
Josko PetricevicDepartment Of Clinical Pathology, Cytology And Forensic Medicine, University Clinical Hospital Mostar, Mostar, Bosnia And Herzegovina
Tel:+387 36342954,
Email:josko.petricevic@yahoo.com
Abstract
A newborn with rare FATCO (Fibular Aplasia, Tibial Campomelia and Oligodactyly) syndrome is described. Syndrome consists of shortening and anterior bowing of the lower limbs at the distal third of tibia, oligodactyly of both feet and fibular aplasia. The infant had no other anomalies. Karyotype was normal male (46 X,Y) and there was no mutation on WNT7A gene. To the best of our knowledge, this is the first described case with bilateral appearance of malformations.
Keywords
FATCO; Fibular aplasia; Oligodactyly; Tibial campomelia
INTRODUCTION
Developmental abnormalities and malformations that have cosmetic or functional significance occur in 3% of newborns [1]. Anomalies of the limbs are rather rare, which is unusual as limb development involves multiple genes [2]. They occur in 1 in 1000 newborn infants and in 2/3 of cases are associated with other malformations [3,4]. The long bone most commonly associated with congenital absence is fibula [4]. Fibular aplasia is often associated with several different syndromes like Du Pan, FFU (Femur-Fibula-Ulna) syndrome, FATCO and Fuhrmann syndrome [4]. Oligodactyly and it is more severe form, ectrodactyly, occur in about 1 birth in 10-18000 and often come with some other abnormalities like Tibial Aplasia Ectrodactyly syndrome (TAE), Holt-Oram syndrome, Poland syndrome, clefting syndrome and ulnar ray syndrome [5-7]. Tibial campomelia, anterior bowing of tibia, is often connected with several syndromes like Campomelic dysplasia and FATCO syndrome [4,5].
Some of these syndromes have genetic basis like Fuhrmann syndrome (WNT7A mutation located on the 3p25, gene that is member of WNT family that encodes signalling proteins included in developmental processes during embryogenesis and oncogenesis), campomelic dysplasia (17q, SOX9 gene mutation), Holt-Oram syndrome (TBX5 mutation), Ectrodactyly-Ectodermal Dysplasia-Clefting syndrome (7q11.2-q21.3 mutation) [4,6].
We report a case of a male newborn with a rarely described congenital limb deficiency syndrome consisting of fibular aplasia, anterior bowing of both lower limbs at the distal third of the tibia with associated overlying soft tissue dimpling and oligodactyly of both feet. It is one of few cases of this syndrome in the world, but it is the first time malformations appeared bilaterally.
CASE
A newborn male was born vaginally after 23 weeks of gestation following fetal distress. The birth mass was 700 grams, length 32 cm, and APGAR score 2. It was immature, bradicardic, atonic, showed no reflexes and was not breathing. Physical examination revealed deformed tibia which were angulated in the distal thirds and olygodactyly on both feet (second and third toe were fused in figure 1). The infant had neither facial dysmorphia nor other associated anomalies (Figure 1). Mild syndactyly of the fourth and fifth digit was visible on the right hand. Blood was taken for cytogenetic analysis, which showed normal male karyotype. Radiographs confirmed tibial campomelia and oligodactyly with four metatarsal bones on each foot (Figure 2). Also, it was evident that both fibulas were missing. Left lung had a discrete shadow while right was without significant pathological findings. The newborn died 6 days after the birth.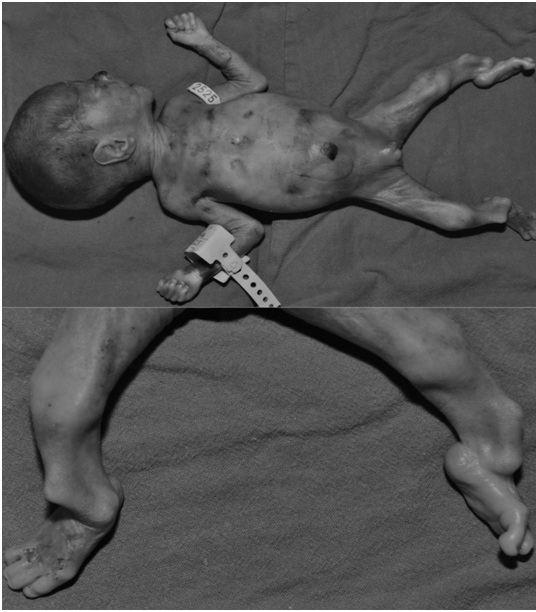
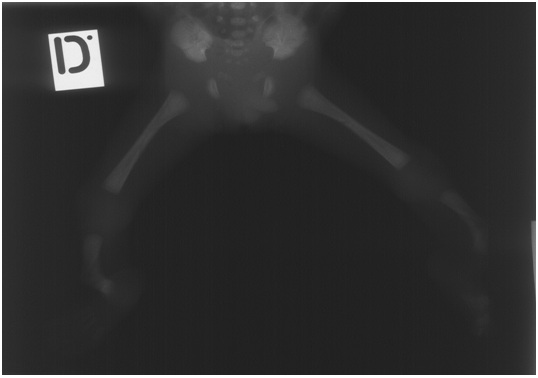
Autopsy showed no other significant pathological findings. Tissue was tested for mutation in WNT7A gene in the DNA extracted from a paraffin block, but this could not be confirmed.
DISCUSSION
There have been only few reports in the literature on the FATCO syndrome, but the case we present is the first report of bilateral malformations. It consisted of fibular aplasia and tibial campomelia of both legs and oligodactyly of both feet. The name was first proposed by Courtens et al., in 2005 [4]. Diagnosis was based mainly on the locations and types of malformations. As previously mentioned there are several syndromes with similar clinical appearance that can be used in differential diagnosis. Table 1 shows these conditions and their clinical appearance.
| Du Pann | Fibular aplasia, brachydactyly [8]. |
| FFU | Amelia, peromelia of humerus, humeroradialsynostosis and defect of ulna [9]. |
| FATCO | Fibular aplasia, tibialcampomelia, oligodactyly [3,4,6]. |
| Fuhrmann | Fibular aplasia/hypoplasia, femoral campomelia, poly/syn/oligodactyly [4]. |
| TAE | Tibial and fibular a/hypoplasia, ectrodactyly, syndactyly [7]. |
| Holt-Oram | Long bones aplasia, carpal bone deformities, ASD, VSD [10]. |
| Poland | Pectoralis muscle aplasia, syn/oligodactyly, ulna/humerus aplasia [10]. |
| Clefting syndrome | Split hand, split foot, ectrodactyly [5]. |
| Campomelic dysplasia | Micrognathia, long bone bowing, cleft palate [5]. |
Table 1: Differential diagnosis.
Most of them are excluded as being this condition due to some differences. FFU syndrome was excluded as femur was not shortened and ulna was normal. Du Pann syndrome (also known as acromesomelic dysplasia) is, beside fibular aplasia, characterised also by brachydactily [8]. Fuhrmann syndrome and one described by Pfeiffer et al., was excluded because these conditions are characterised by femoral bowing, not tibial [4]. Also, Fuhrmann was excluded by analysis of WNT7A gene. This gene shows no mutations in FATCO syndrome which has the exact clinical appearance as the one described in the case with one exception [4].
The sex ratio in all described cases appears to be biased toward males. To the best of our knowledge, there were only two reports of FATCO syndrome in girls. Other cases with affected females had similar but not the same types of malformations. Some had femoral bowing or polidactyly but only Hecht et al., and Lazjuk et al., described this syndrome in female patients [11,12].
Also, several reported patients have other malformations beside described three - fibular aplasia, tibial campomelia and olygodactyly [11-13]. Whether this represents a variability in the same syndrome or appearance of other malformations beside FATCO syndrome needs to be ascertained.
There are only eight reported cases of FATCO syndrome available in the literature (search of PubMed using the keywords “FATCO, fibular aplasia, tibial campomelia, oligodactyly”) but no reports on its bilateral appearance. Huber et al., described partial bilaterality of similar malformations in their case 2, where the patient had bilateral tibial campomelia and fibular aplasia with oligodactyly of the hand [14]. However, they categorised the case as Fuhrmann syndrome although Fuhrmann has femoral but not tibial bowing [14]. Kitaoka et al., reported bilateral oligodactyly and tibial campomelia but unilateral fibular aplasia [13]. Our case it the first case of proven FATCO syndrome with bilateral malformations. It is still unclear what would influence unilateral or bilateral appearance as there are only eight reported cases.
There are several genes that participate in leg development but no genetic basis of FATCO syndrome has been identified so far. The cluster of homeobox D genes located on chromosome 2 is involved in limb development [15]. They act as sequence-specific transcription factors expressed in the developing limb buds and have been implicated in severe limb and genital abnormalities. Our patient at 23 weeks of gestation is the youngest patient described, so it is reasonable to conclude that malformation occurs before this age. Future genetic studies should address the developmental aspect of FATCO syndrome and provide insight into the development of human lower limbs.
REFERENCES
- Kumar V, Abbas AK, Fausto N (2005) Robbins and Cotran’s Pathologic Basis of Disease (7thedn). Elsevier Saunders, Philadelphia, USA.
- Ekbote AV, Danda S (2012) A Case Report of Fibular Aplasia, Tibial Campomelia, and Oligosyndactyly (FATCO) Syndrome Associated With Klinefelter Syndrome and Review of the Literature. Foot Ankle Spec 5: 37-40.
- Vyskocil V, Dortova E, Dort J, Chudacek Z (2011) FATCO syndrome - fibular aplasia, tibial campomelia and oligosyndactyly. Joint Bone Spine 78: 217-218.
- Courtens W, Jespers A, Harrewijn I, Puylaert D, Vanhoenacker F (2005) Fibular aplasia, tibial campomelia and oligosyndactyly in a male newborn infant: a case report and review of the literature. Am J Med Genet A 134: 321-325.
- Jones K, Smith DW (2005) Smith’s recognizable patterns of human deformation (6thedn). Elsevier Saunders, Philadelphia, USA.
- Karaman A, Kahveci H (2010) A male newborn with FATCO syndrome (fibular aplasia, tibial campomelia and olygodactyly): A case report. Gen Couns 21: 285-288.
- Mohammed N, Mahmoud TA, Sabitha K (2006) Ectrodactyly with aplasia of long bones (OMIM; 119100) in a large inbred Arab family with an apparent autosomal dominant inheritance and reduced penetrance: Clinical and genetic analysis. Am J Med Genet A 140: 1440-1446.
- Al Kaissi A, Ghachem MB, Chehida FB, Kozlowski K (2005) Acromesomelic dysplasia du Pan. Magyar Radiologia 79: 234-239.
- Geniets C, Vanhoenacker F, Blaumeiser B, Parizel PM (2006) Femur-fibula-ulna complex. JBR-BTR, 89: 130-131.
- Pedijatrija MD (2003) Školska knjiga, (7thedn). Zagreb, Croatia.
- Hecht JT, Scott CI Jr (1981) Limb deficiency syndrome in half-sibs. Clin Genet 20: 432-437.
- Lazjuk JI, Lurie IW, Cherstvoy ED, Ussova YI (1976) A Syndrome of multiple congenital malformations including amelia and oligodactyly occurring in half cousins. Teratology 13: 161-165.
- Kitaoka T, Namba N, Kim JY, Kubota T, Miura K, et al. (2009) A Japanese male patient with ‘fibular aplasia, tibial campomelia and oligodactyly’: An additional case report. Clin Pediatr Endocrinol 18: 81-86.
- Huber J, Volpon JB, Ramos ES (2003) Fuhrmann syndrome: Two Brazilian cases. Clin Dysmorphol 12: 85-88.
- Bittar EE, Bittar N (1998) Developmental biology. JAI Press, Greenwich, Connecticut, USA.
Citation: Petricevic J, Curic A, Karaman I, Forempoher G, Definis-Gojanovi? M (2017) First Case of Bilateral Fibular Aplasia, Tibial Campomelia and Oligodactyly Syndrome (FATCO syndrome). J Clin Stud Med Case Rep 4: 046.
Copyright: © 2017 Josko Petricevic, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.