Frequency and Anatomical Fetchers of Tricuspid Valve Atresia in Azerbaijan: Based On Fetal Cardiology Findings
*Corresponding Author(s):
Petropoulos ACDepartment Of Icu Cardiac Neonatal Pediatric, Great Ormond Street Hospital, London, United Kingdom
Tel:+44 7444251425,
Email:Andreas.Petropoulos@gosh.nhs.uk
Abstract
Aim
To describe the incidence, the prenatal diagnosis and the strategy of treatment of patients suffering from Tricuspid Valve Atresia (Tv atr.).
Methods
We describe 10 cases of prenatal diagnosis from our series of our fetal cardiac clinic in three different hospitals, during the period 2012-18 from Baku-Azerbaijan. We review the existing literature on the pre-natal diagnosis and post-natal management.
Results
We present the key findings for diagnosis by fetal echocardiography that include: Demonstration of no patent tricuspid valve on the 4-Chamber (4-ch) view, no flow across the Tv on pulse or color Doppler flow mapping, small Right Ventricle (RV) with associated Ventricular Septal Defect (VSD) and the Great Vessels (GA’s) connectivity to the ventricles (normal related or transposed). The abnormal 4ch view RV, gives the primary clue leading to follow-up series of scans that can determine the immediate post-natal anatomy and further management of the patient.
Conclusion
Tv atr. can be accurately diagnosed prenatally. The key findings, differential diagnoses and management are discussed. Short review of the literature of the disease is provided.
Keywords
Fetal Echocardiography; Prenatal diagnosis; Strategies in treating tricuspid valve atresia; Tricuspid valve atresia
ABBREVIATIONS
Tv atr.: Tricuspid Valve Atresia
4ch: Four Chamber
CHD: Congenital Heart Defect
HRHS: Hypoplastic Right Heart Syndrome
HLHS: Hypoplastic Left Heart Syndrome
RV: Right Ventricular
RA: Right Atrium
Tv: Tricuspid Valve
d-TGA: D- Transposition of Great Arteries
ToF: Tetralogy of Fallot
LA: Left Atrium
ASD II: Secundum type Atrial Septal Defect
PFO: Paten Foramen Obale
AVv: Atrioventricular Valve
Mv: Mitral Valve
VSD: Ventricular Septal Defect
CoA: Aortic Coarctation
PDA: Patent Ductus Arteriosus
GA’s: Great Arteries
TA: Trancus arteriosus
DORV: Double Outlet RV
DOLV: Double Outlet LV
PAvS: Pulmonary valval stenosis
Qp /Qs: Pulmonary-to-Systemic blood flow ratio
LVvo: LV Volume Overloading
SVD: Spontaneous Vaginal Delivery
PGE: Alprostadil
BT-Shunt: Blalock-Taussig Shunt
TCPC: Total Cavopulmonary Anastomosis
INTERDICTION
Tricuspid Valve Atresia (Tv atr.) is a form of a Hypoplastic Right Heart Syndrome (HRHS). The defect was firstly published in 1817 by Kreysig. The essence of the malformation lies in the absence of a functional tricuspid valve between the Right Atrial (RA) and the Right Ventricle (RV) [1].
EPIDEMIOLOGY
In our series, Tv atr., is described as the third most common cyanotic Congenital Heart Defect (CHD). From 774 fetal scans, (Petropoulos AC et al., unpublished data), was the second most common, after Tetralogy of Fallot (ToF) [2]. The other 2 frequently observed were: TGA and DORV. Worldwide prevalence is reported between 1- 2.3% of all CHD [2] (Table 1). Our incidence was 2.7 times higher than the reported worldwide. We believe that the spectrum of HRHS is higher in our area due to a possible endogenous genotype like those reported up to date [3]. This can probable be explained as in Azerbaijan the incidence of CHD detected among term neonates in the country is high 1.82% [4].
Table 1: Geographic Prevalence of Tv atr.
ANATOMICAL FEATURES
Most common histological type of Tv atr., is the muscular, found in 89% of cases. Localized fibrous thickening in the site of the Tv [5] can be found (Figure 1). Rare types are: membranous (6.6%), in which the atrioventricular part of the membranous septum forming the floor of the Right Atrium (RA) is atretic. This type is associated with absent PAv leaflets. In 2.6% of cases, an Ebstein type, combining the essence of both defects can exist. Approximately 1% the valval cusps are minute and fused. In the atrioventricular canal type, (0.2%) an atretic leaflet of the common Atrioventricular Valve (AVv), guards the inflow to the RV [5]. In the unguarded type (0.6%), the atrioventricular junction is unguarded, but the inlet component of the RV is separated from its outlet by a muscular shelf [5] (Figure 4). In an atretic Tv setting, an intra atrial communication to drain systemic venous return to the Left Atrium (LA) is obligatory. This can either be an unrestrictive secundum type Atrial Septal Defect (ASD II) or even a large or restrictive Paten Foramen Obale (PFO). This will lead to volume overload and finally, hypertrophy of the LV and MvR+. Its severity will correlate with any delay in surgical repair. An obligatory- in the classic form -VSD is present allowing the blood to enter the hypoplastic RV that size and function very, regarded to the relationship of the GA’s [5]. The VSD may be: Cono-ventricular, perimembranus, cono-septal mal-aligned, muscular or atrioventricular canal type [5]. From these the muscular is most frequent and can restrict by time. Subpulmonary stenos is in patients with normally related GA’s and simulate sub-aortic obstruction in patients with transposition of the GA’s can be found [6]. GA’s relationship is variable and forms the basis of classification [7]. Aorta is either normal or slightly larger. Obstruction to the pulmonary outflow tract is frequent [7].
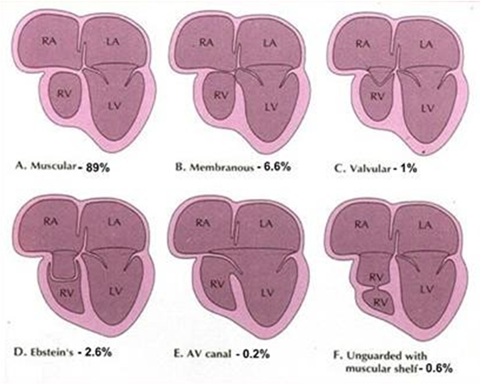 Figure 1: Anatomical Variations of Atretic Tricuspid Valve.
Figure 1: Anatomical Variations of Atretic Tricuspid Valve.
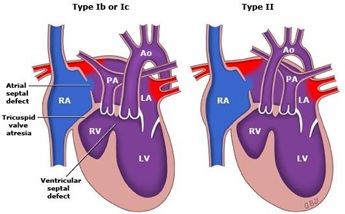 Figure 2: Most common types of Tv atr.
Figure 2: Most common types of Tv atr.
In 30% of patients, various associated cardiac defects are present, with aortic Coarctation (CoA) and persistent left superior vena cava, most common. These can be:
- Interrupted aortic arch.
- Absent PAv.
- Aneurysm of the atrial septum.
- Aorto-Pulmonary fistula.
- Right aortic
- Anomalous origin of the coronary arteries from the pulmonary artery (ALCAPA).
- Anomalous origin of the left and or right subclavian artery.
- Common atrium.
- Cor triatriatum dexter type.
- Coronary sinus atrial septal
- Double aortic arch.
- Hemi and persistent Truncus Arteriosus [8].
- Hypoplastic ascending aorta and/or
- Atresia Primum (ASD).
- Uhl’s heart.
- Patent Ductus Arteriosus (PDA).
- Total anomalous pulmonary venous drainage.
- Tubular hypoplasia of the aortic arch.
- Aortic valve stenosis.
- Isomerism atrial appendages
- Anomalous drainage of coronary sinus into LA [9] Associated extra cardiac congenital malformations are reported in 22%. Most common: Chromosomal anomalies with frequent VACTERL association, unilateral renal agenesis, hypospadias, hydrothorax, megacystis and agenesis of the ductus venosus [10,11].
CLASSIFICATION OF THE DEFECT
Many classifications exist. The most commonly used is based on the interrelations of the GA’s and the amount of PAv Stenosis (PAvS) that may exist [7].
- Type I Normally related GA’s (~70%) (Figure 2).
- Type II D-Transposition of the GA’s (~29%) (Figure 2).
- Type III GA’s positional abnormalities other than D-transposition: a. Subtype-1: L-transposition, b. subtype-2: DORV, subtype-3: DOLV, d. subtype-4: D-malposition of GA’s, e. subtype-5: L-malposition of GA’s.
- Type IV TA [7,10].
- Subgroup a: With PAv atresia.
- Subgroup b: With PAvS or hypoplasia.
- Subgroup c: Without PAvS.
Further, the status of VSD-patent or not-and the presence of other associated malformations are described.
This unified classification [7] includes all types described by previous classifications by Kuhne, Edwards and Burchell, and by Keith, Rowe and Vlad [12].
EMBRYOLOGY
The AVv’s develop shortly after the atrioventricular cushions are activated. The anterior and posterior Tv leaflets develop by undermining of a skirt of ventricular muscle tissue. The septal leaflet mostly develops from the inferior endocardial with a small contribution from the superior cushion. The process of undermining extends until the AVv junction is reached. Resorption of the muscle tissue produces normal-appearing valve leaflets and hordae tendineae. Fusion of developing valve leaflet components results in stenosis or atresia [13]. Whether a muscular type or a fused valve develops depends on the embryologic stage in which aberration takes place [6,14] (Figure 1).
The embryologic, pathologic, clinical, and imaging features of Tv stenosis and/or atresia are similar. This leads rare seen Tv stenosis, to be calcified in this group of CHD [14,15].
PATHOPHYSIOLOGY
During the fetal period, systemic venous blood return is forced across the PFO into the left heart. The lowered PaO2 to the brain and heart and elevated PaO2 to the lungs do not produce postnatal abnormalities [7]. In patients with type I and associated PAv atresia types Ia and IIa, their pulmonary blood flow is supplied entirely through the PDA. This shunt delivers 8-10% of combined ventricular output compared with 66% of combined ventricular output in a normally developed fetus. Acute angulations of the PDA occurs, because of reversed direction of DA flow. These two factors make the PDA less responsive to postnatal stimuli than usual.
In a fetus with type I anatomy and a small or absent VSD, almost all LV output is ejected into the aorta and transported to the placenta. Therefore, as the aortic isthmus carries a larger-than-normal cardiac output CoA is rarely found in this subtype.
Fetuses with type II have increased portion of the blood entering by DA into the descending aorta. This results minimal flow across the aortic isthmus. As a result, this can lead to frequent seen CoA [1,16].
Postnatally, the obligatory PFO/ASDII causes a mixing of systemic and pulmonary venous returns, in the LA and passes into the LV [6-7,16]. This flow pattern occurs in all types of Tv atr. except of type III and subtypes 1 and 5. In these exceptions, the atretic Tv is left sided due to ventricular inversion; therefore, the pathophysiology is that of Mv atresia with consequent left-to-right shunting of pulmonary venous return [6,16].
In patients with normally related GA’s, and a VSD, shunting through the VSD permits perfusion of the lungs. When the VSD is absent, pulmonary blood flow is derived by the PDA or aorto-pulmonary collateral vessels [6-7,16].
In patients with D-TGA’s, the lungs receive blood flow from the LV. The aorta receives blood from the LV via the VSD and RV [6-7,16]. In other types, the routes of aortic and pulmonary artery flow depend on the size of the VSD and associated cardiac defects.
Additional conditions that affect the clinical presentation and treatment plan are:
ARTERIAL DESATURATION
An amount of systemic desaturation is present in all patients, due to obligatory mixture of systemic, coronary, and pulmonary venous returns, in LA. The degree of arterial desaturation depends on the amount of pulmonary blood- flow [7,16]. The arterial oxygen saturation has a curvilinear relationship, with a pulmonary-to-systemic blood flow ratio (Qp /Qs) that reflects the pulmonary blood flow. Qp/Qs ratio of 1.5-2.0 results in adequate oxygen saturation. Higher Qp does not significantly increase saturation and produces LVvo and pulmonary over-circulation, with tachypnoea that can lead to CHD and pulmonary edema [7,16].
Pulmonary blood flow
Clinically, patients depend mainly on the quantity of Qp [7,16]. Neonates with markedly decreased Qp are likely to present early with severe cyanosis, hypoxemia, acidosis. When Qp is increased, the neonates may appear mild cyanotic but show signs of heart failure later. Patients with pulmonary oligemia generally have type I disease. Those with pulmonary plethora usually have type II and, rarely, type I c [7,16].
In patients with a type I defect, the obstruction can be valvar, sub valvar, or frequently, at the VSD level. In patients with a type II defect, the obstruction is valvar or sub valvar. In patients with a type I defect, with large VSD, without PAvS, the Qp is inversely proportional to the pulmonary-to-systemic vascular resistance ratio. If a PDA or a surgical systemic-to-pulmonary artery shunt has performed, Qp, is proportional to the size of the natural or surgical shunt [7,16].
LV volume overloading (LVvo)
LV ejects the entire outputs. Therefore, LVvo is present in all patients [6,16]. The degree of LVvo increases further if mild or absent pulmonary outflow obstruction is noted or if systemic-to-pulmonary artery shunting was performed. Maintaining normal LV function is essential for a successful Fontan operation. LV function tends to decrease as age increases, decreasing Qp/Qs, and arterial desaturation [16,17].
Obstruction of the interatrial communication
PFO/ASD II, is essential for survival. As entire systemic venous blood must pass through, any obstruction if present can be clinically significant [6,16]. PFO/ASD II obstruction, is present when the mean pressure difference between RA-LA is more than 5 mm Hg [1,7] and/or the diameter of the defect is less than 5 mm [1,7].
FINDINGS
In our series we detected ten cases of Tv atr. These were six of type I from which four was subgroup b. One was subgroup a and one subgroup c. Three cases were type II. Two were subgroup b and one subgroup c. Finally, one case was type III - subtype-2 and subgroup b. So, it could be also called DORV with Tv atr. and PAv st. The indications for fetal cardiology evaluation were: 1. Inbreeding (first- and second-degree relatives among parents) 3/10 (30%) 2. Abnormal cardiac screening examination during the 20th week anomaly scans 2/10 (20%) 3. IVF conceived fetuses, 2/10 (20%), 4. First-degree relative of a fetus with congenital heart disease1/10 (10%), 6. Combination of maternal age and increased nuchal translucency 1/10(10%) 7. Maternal Metabolic disorder (Diabetes Mellitus) 1/10(10%).
The initial scans were done mostly after the 20th week of gestation, with a mean gestational age (21+/-3 weeks). The assessment was done using color Doppler -2D echocardiography, by a General Electric Vivid 7 device, with an appropriate cardiac fetal probe and software program.
The findings were verified with echocardiography, cardiac catheterization, cardiac CT and MRI studies were indicated, post-natal, and postmortem examination in pregnancies that were interrupted.
Post-natal 4 female and 2 male neonates were delivered by cesarean section between 36-38th week of gestation in centers that had Neonatal Intensive Care Units. Their anatomy was in all cases type I. Four were subgroup b and the other two were subgroup’s a and c.
All families were thoroughly consulted on two consecutive meetings on findings and further management and outcomes that could be offered locally or abroad.
From the ten cases four 4/10(40%) choose to terminate the pregnancy. These were 3/4 (75%) type II and one (25%) was type III - subtype-2 and subgroup b.
The interruption of pregnancy took place between 24 +/-2 weeks of gestation. Interruption of pregnancy due to significant medical issues in legal in Azerbaijan before the 28th week of gestation. The post-mortem revealed two females’ fetuses and two males. No macroscopic additional malformations were noticed. The cardiac anatomy was identical to the prenatal findings.
From the delivered babies four received prostin during the first day of life and was later offered an early Blalock-Taussig shunt at age 50+/-6 days old. One type I-subgroup C presented at age 35 days old with symptoms of CHF. She was palliated with a band of her main Pulmonary artery.
One female type I-subgroup b had balanced physiology and did not require any initial stage of palliative surgery or prostin [1].
DISCUSSION
Tv atr. is a rather rare prenatal diagnosed CHD. In our series it was the third most common cyanotic CHD [2]. Probable this was due to the cumulation of possible pathological cases as we are the referral center for fetal cardiology in the country. The true incidence of the defect will be determined only by a prospective epidemiology study among living neonates [4].
Tv atr. as a defect in the spectrum of HRHS must be linked to an abnormal 4-chamber view and therefore, it must be potentially detectable during routine obstetric sonographic scans [18].
Fetal Cardiology diagnosis must emphasize on:
- No patency of Tv. This can be seen by 2D and color Doppler (Figure 3) [11,19].
- Cardiac axis, position, cardiothoracic ratio and situs of the viscera are normal.
- Discrepancy between RV and LV is always present.
- MvR+ is frequent and due to LVvo. [19] (Figure 4).
- Nearby 70% have normal related GA’s, frequently with asymmetry in the PAv/Aov ratio. Approximately 29% have D-TGA [11] (Figure 2).
- Aortic arch in most cases is normal. The ductal arch is larger than normally, with a reversed ductal flow [19].
- Moderate at least size VSD with a left to right shunt is frequently present [11] (Figure 4).
- Rarely restrictive right to left flow through the FO [19].
- The LV has a normal function and no evidence of endocardial fibro elastosis is seen in undeveloped RV [17,19].
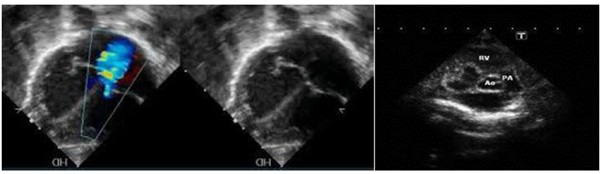 Figure 3: a) absence of color flow mapping through the atretic tricuspid valve and regurgitate jet through the dilated Left Ventricular; b) Fetal echocardiography, colored Doppler showing no patency of the atretic tricuspid valve b) Fetal echocardiography showing c) Fetal echocardiography showing common type I tricuspid valve atresia.
Figure 3: a) absence of color flow mapping through the atretic tricuspid valve and regurgitate jet through the dilated Left Ventricular; b) Fetal echocardiography, colored Doppler showing no patency of the atretic tricuspid valve b) Fetal echocardiography showing c) Fetal echocardiography showing common type I tricuspid valve atresia.
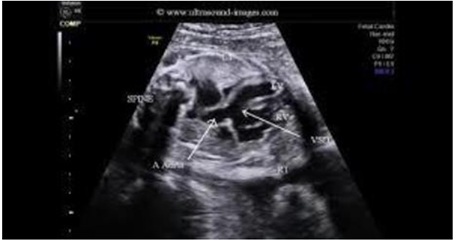 Figure 4: Fetal echocardiography showing VSD setting, hypoplastic RV in the setting of tricuspid valve atresia.
Figure 4: Fetal echocardiography showing VSD setting, hypoplastic RV in the setting of tricuspid valve atresia.
When detected prenatally, a 4-week follow-up during the pregnancy is suggested to verify the final anatomy. As extra cardiac malformations and chromosomal abnormalities is low amniocentesis is not recommended [18]. The risk of recurrence is only 1-2% in a next pregnancy [18,20]. Most are sporadic although familial cases have been reported [21].
If no prenatal data exist, postnatal diagnosis can be achieved by the failing pulse oximetry screening test [22,23]. The clinical presentation will be based on the existing anatomy and physiology. If oxygen saturation is less than 70-75%, an intervention to improve pulmonary oligemia must be considered. Electrocardiogram is diagnostic, revealing RA dilatation, abnormal superiorly oriented QRS axis, LVH, and decreased RV forces. RA dilatation, with tall and peaked P waves (≥2.5 mm) in lead D-II and right chest leads are present in 75% of patients. The so-called P-tricuspidale with a double peak, spike and dome configuration may be present. Abnormal vector is present in 80% of patients with Tv atr., type I but only in 50% of patients with type II or III [24]. Echo-2D will establish the diagnosis and verify all the pathology, in most of the cases. Reveals a small RV and an enlarged RA, LA and LV. In commonest muscular type, a dense band of echoes is observed in the Tv location. The anterior leaflet of the detectable AVv is attached to the left side of the interatrial septum. These features are best demonstrated in the apical and subcostal 4-ch views. Imaging of ASD and VSD are essential. CoA, more frequent in patients with type II, will be showed in the suprasternal view [25].
Cardiac catheterization that was so commonly used, in the era of sophisticated Echo-2D and cardiac MRI is nowadays, limited. A few cases in which specific hemodynamic data are required, verification of some of the Choussat criteria for planning surgical repair or a combination of diagnostic and interventional procedures when needed, will enter the Cath Lab [20]. An existing CoA can be addressed initially by an interventional procedure [2,22,26-27]. Finally, interventional procedures can be used following the surgical repair to address residual conditions.
MEDICINE TREATMENT
Neonates with severe cyanosis, pulmonary oligemia and PDA-dependent pulmonary flow can benefit by intravenous administration of alprostadil (PGE1). PGE1 is effective only in the neonatal period. A modified or classical Blalock-Taussig Shunt (BT-Shunt) should be offered to improve cyanosis.
If congestive heart failure is present, diuretics and after load-reducing agents as Angiotensin-converting enzyme inhibitors may be administered to reduce after load and augment LV function, especially after a bidirectional Glenn procedure and a Fontan type operation. Anticoagulants and platelet-inhibiting drugs are useful in preventing thrombus formation after a Glenn or a Fontan completion [20,27].
The American Heart Association recommends that prophylaxis with antibiotics be given before any bacteremia-producing procedures are performed [20,27].
SURGICAL TREATMENT
The patient’s age, weight, anatomic and physiologic status determines the types of surgery recommended. The overall objective is to achieve a Fontan circulation by a total cavopulmonary connection [20].
In neonates and young infants with pulmonary oligemia, as a first step, a classic or modified BT-shunt is undertaken to improve oxygenation [27].
In patients aged 6 months to 2 years, a bidirectional Glenn procedure (Figure 5) is the goal standard approach. For patients older than 2 years, total cavopulmonary connection may be performed, but most authorities suggest staging by using an initial bidirectional Glenn operation followed by Fontan completion after 6-12 months, or even after the age of 4 years old [20,27].
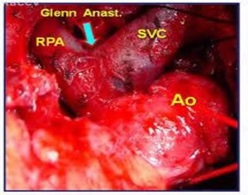 Figure 5: Surgical illustration of a Bi- Directional glenn anastomosis.
Figure 5: Surgical illustration of a Bi- Directional glenn anastomosis.
During bidirectional Glenn surgery, any narrowing of the pulmonary artery should be repaired. Other additional existing lesions such as sub-aortic obstruction and MvR+ should also be addressed [20,27]. Before Fontan completion, especially in delayed presenting or poorly followed up cases, some suggest cardiac catheterization to be undertaken. This approach is widely abandoned as MRI studies become more frequently used. A cardiac catheterization or an MRI study needs to investigate the possibility of existing aorto-pulmonary collaterals. If collateral vessels are present, they should be occluded with coils prior to the Fontan Completion [20,26-27].
Most surgeons currently prefer extra-cardiac conduit diversion of inferior vena cava blood into the right pulmonary artery. To address the growth issue related to extra-cardiac Fontan surgery, some surgeons use autologous or bovine pericardial roll grafts [20,27].
In patients with D-TGA’s, early banding of the PA, relief of possible CoA and bypassing using a Daimus-Kaye- Stansel procedure and/or resecting if technically possible of the sub-aortic obstruction should be incorporated into the management plan [20,27,28].
Following the Fontan completion, immediate, mid and long-term complications that can occur are: Persistent pleural effusions, neurologic complications such as strokes or brain abscesses, arrhythmias, thromboembolic events, obstruction at the anastomotic sites or in the pulmonary artery, residual shunts and/or systemic venous congestion presenting as protein-losing enteropathy and rarely plastic bronchitis. All these need to be aggressively treated to sustain a New York Heart Association stage I to II and improve quality and life expectancy [20].
In patients with “Failing Fontan” after excluding/addressing obstructions, residual shunts and arrhythmias, apart from conventional treatment, consideration for resynchronization by pacing the RA and LV and conversion of
atrioventricular Fontan’s to Total Cavopulmonary Anastomosis (TCPC) (Figure 6) Cardiac transplantation should be considered as the last step of further treatment in a failing Fontan circulation [20,27,29].
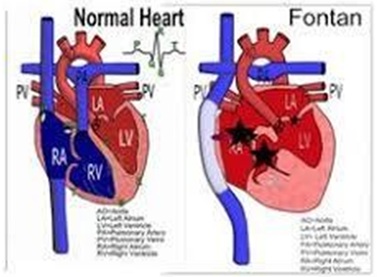 Figure 6: Fontan Circulation after TCPC in palliation of Tv atresia.
Figure 6: Fontan Circulation after TCPC in palliation of Tv atresia.
PROGNOSIS
Prognosis in untreated Tv atr. is poor. Early identification, rapid and safe transport to a pediatric cardiology center, noninvasive diagnosis, availability of PGE1 to keep PDA, and recent advances in anesthesia and surgical techniques have improved the prognosis of these babies. Complications associated with the classic Fontan are decreased with the widespread use of staged TCPC (Figure 6) [20,27-28].
CONCLUSION
Tv atr. is a complex cyanotic critical CHD requiring multiple medical, intervention and surgical treatment. Prenatal diagnosis is available alternating early morbidity and mortality but also offers to parent the alternative of an elective abortion. A detailed explanation of the cardiac defect and treatment required should be given to the parents at the time of diagnosis and repeated as needed [20,27]. Further, pulse oximetry screening after the 3ed postnatal day can detect this CHD [22].
CONFLICT OF INTEREST
None of the authors have any conflict of interest in this series of Tv atr.
REFERENCES
- Petropoulos AC, Xudiyeva A, Valiyeva Q, Behbudov V, Ismaylova M (2018) Prenatal diagnosis and management of tricuspid valve atresia: A case report and review of literature. J Neonatol Clin Pediatr 5: 1-7.
- Petropoulos AC, Xudiyeva A, Valiyeva Q, Behbudov V, Maharammova T, et al. (2015) Incidence of congenital cardiac disease, detected by fetal echocardiography, in experience of three centers during a thirty month period. Card in the Young 25: 1.
- Dimopoulos A, Sisko RJ, Kay DM, Rigler SL, Druschel CM, et al. (2017) Rare copy number variants in a population based investigation of hypoplastic right heart Birth Defects Res109: 8-15.
- Petropoulos AC, Hudiyeva A, Behbudov V, Mustafayeva G, Guliyev N, et al. (2019) The Incidence of Congenital Heart Disease in Baku-Azerbaijan. Prospective Epidemiology Study. Arch Dis Child-epa 104: 1-428.
- Rao PS (1992) Terminology: Is tricuspid atresia the correct term to use? In: Rao PS (eds.). Tricuspid Atresia, (2nd edn). Futura, Mount Kisco, Pg no: 3-15.
- Ottenkamp J, Wenink AC, Quaegebeur JM, Rohmer J, Gittenberger-de Groot AC, et al. (1985) Tricuspid atresia. Morphology of the outlet chamber with special emphasis on surgical implications. J Thorac Cardiovasc Surg 89: 597-603.
- Rao PS (1990) Tricuspid atresia. In: Long WA (eds.) Fetal and Neonatal Cardiology. WB Saunders, Philadelphia, Pennsylvania, USA. Pg no: 525-40.
- Rudolph AM (1974) Tricuspid atresia with hypoplastic right Congenital Disease of the Heart. Year Book Medical, Chicago, Illinois, USA. Pg no: 429-61
- Rao PS, Levy JM, Nikicicz E, Gilbert-Barness EF (1991) Tricuspid atresia: Association with persistent truncus Am Heart J 122: 829-835.
- Rao PS, Covitz W, Chopra PS (1992) Principles of palliative management of patients with tricuspid In: Rao PS (eds.). Tricuspid Atresia, (2nd eds.). Futura, Mount Kisco, NY, USA. Pg no: 297-320.
- Berg C, Lachmann R, Kaiser C, Koiowski P, Stressig R, et al. (2010) Prenatal diagnosis of tricuspid atresia: Intrauterine course and outcome. Ultrasound Obstet Gynecol 35: 183-190.
- Rao PS (1980) A unified classification for tricuspid atresia. Am Heart J 99: 799-804.
- Van Mierop LH, Gessner IH (1972) Pathogenetic mechanisms in congenital cardiovascular Prog Cardiovasc Dis 15: 67-85.
- Keefe JF, Wolk MJ, Levine HJ (1970) Isolated tricuspid valvular stenosis. Am J Cardiol 25: 252-257.
- Lang D, Oberhoffer R, Cook A, Sharland G, Allan L, et al. (1991) Pathologic Spectrum of Malformations of the Tricuspid Valve in Prenatal and Neonatal Life. J Am Coll Cardiol 17: 1161-1167.
- Rao PS (1991) Perinatal circulatory physiology. Indian J Pediatr 58: 441-451.
- Graham TP Jr, Erath HG Jr, Boucek RJ Jr, Boerth RC (1980) Left ventricular function in cyanotic congenital heart Am J Cardiol 45: 1231-1236.
- Tongsong T, Sittiwangkul R, Wanapirak C, Chanprapaph P (2004) Prenatal diagnosis of isolated Tricuspid valve atresia: Report of 4 cases and review of the literature, J Ultrasound Med 23: 945-950.
- Bader RS, Hornberger LK, Huhta JC (2008) Tricuspid Atresia in The Perinatal Cardiology Mobile Medicine Series, USA.
- Kanakis MA, Petropoulos AC, Mitropoulos FA (2009) Fontan operation. Hellenic J Cardiol 50: 133-141.
- Tzifa A, Gauvreau K, Geggel RL (2007) Factors associated with development of atrial septal restriction in patients with tricuspid atresia involving the right-sided atrioventricular valve. Am Heart J 154: 1235-1241.
- Petropoulos AC (2018) Right use of Pulse Oximetry must be used as a Screening Test for early detection of critical Congenital Heard Diseases. Open J Cardiol Heart Dis 1-3.
- Mahle WT, Newburger JW, Matherne GP, Smith FC, Hoke TR, et al. (2009) Role of Pulse Oximetry in Examining Newborns for Congenital Heart Disease: A Scientific Statement from the AHA and AAP. Pediatric 124: 823-836.
- Gamboa R, Gersony WM, Nadas A (1996) The Electrocardiogram in Tricuspid Atresia and Pulmonary Atresia with Intact Ventricular Septum. Circulation 34: 24-37.
- Covitz W, Rao PS (1992) Non-invasive evaluation of patients with tricuspid atresia ((roentgenography, echocardiography and nuclear angiography). In: Rao PS (ed.) Tricuspid Atresia, (2nd edn). Futura, Mount Kisco, NY. Pg no: 165-82.
- Rao PS (1992) Cardiac catheterization in tricuspid In: Rao PS (ed.) Tricuspid Atresia, (2nd edn). Futura, Mount Kisco, NY. Pg no: 153-78.
- Mavroudis C, Backer CL (2003) Pediatric cardiac Surgery, 3rd, Philadelphia, Pennsylvania, USA.
- Rao PS (2007) Principles of management of the neonate with congenital heart disease. Neonatology today 2: 1-10.
- Jayakumar KA, Addonizio LJ, Kichuk-Chrisant MR, Galantowicz ME, Lamour JM, et al. (2004) Cardiac transplantation after the Fontan or Glenn procedure. J Am Coll Cardiol 44: 2065-2072.
Citation: Petropoulos AC, Xudiyeva A, Valiyeva Q, Behbudov V, Ismaylova M (2019) Frequency and Anatomical Fetchers of Tricuspid Valve Atresia in Azerbaijan: Based On Fetal Cardiology Findings. J Neonatol Clin Pediatr 6: 038.
Copyright: © 2019 Petropoulos AC, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.