Fulminant Danon Disease in a Young Female Patient with a Novel LAMP2 Mutation
*Corresponding Author(s):
Eric AdlerUniversity Of California San Diego, San Diego, CA, United States
Tel:+1 8586578530,
Email:eradler@health.ucsd.edu
Abstract
A 14-year-old female presented for congestive heart failure with biventricular hypertrophy and severely depressed LVEF. Cardiac MRI findings of extensive LGE but sparing the mid septum, genetic tests reporting a LAMP2 missense mutation and LAMP2 protein staining in the minority of cardiac myocytesat immunohistochemistry suggested a Danon Disease diagnosis.
History of Presentation and Past Medical History
A 14-year-old female with a history of abdominal pain, gastroesophageal reflux disease, attention-deficit/hyperactivity disorder, anxiety and blurry vision for bilateral retinal pigment epitheliopathy presented to the Emergency Room (ER) with several weeks of worsening exertional dyspnea and lower limb edema.
Upon presentation the patient was alert, with no acute distress, tachycardic (HR 112 bpm) and normotensive (BP 116/57 mmHg). Physical exam was notable for pitting edema bilaterally, reduced lung sound bilaterally and hepatomegaly.
Differential Diagnosis And Investigations
Chest X-ray showed pulmonary edema and cardiomegaly. Laboratory investigations showed, elevated troponin I (1.83 ng/mL), BNP (12139 pg/mL), abnormal liver function (ALT 420 U/L; AST 290 U/L, bilirubin 4.0 mg/dL), and high C-reactive protein (6.74 mg/dL).
The ECG was significant for Right Ventricle (RV) hypertrophy and showed a short PR interval consistent with pre-excitation. She underwent a Transthoracic Echocardiogram (TTE) which confirmed RV hypertrophy with reduced systolic function and showed a dilated and hypertrophic LV (end-diastolic volume 160 ml; Intraventricular septum end-diastole 1.2 cm, z-score 1.6; LV mass 329.8 g, z-score 3.3) with severely depressed ejection fraction (EF 19%) and global hypokinesia (Figure 1, Videos 1,2&3).
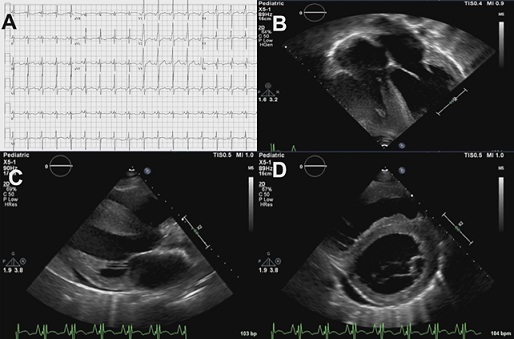 Figure 1: A) Electrocardiogram showing sinus tachycardia (112 beats/min), right ventricular hypertrophy, short PR interval, slurring of QRS complexes upstroke; B,C,D): Cardiac ultrasound showing severe biventricular concentric hypertrophy with LV dilatation and mild pericardial effusion (Online Videos 1, 2,3).
Figure 1: A) Electrocardiogram showing sinus tachycardia (112 beats/min), right ventricular hypertrophy, short PR interval, slurring of QRS complexes upstroke; B,C,D): Cardiac ultrasound showing severe biventricular concentric hypertrophy with LV dilatation and mild pericardial effusion (Online Videos 1, 2,3).
https://www.heraldopenaccess.us/fulltext/Clinical-Studies-&-Medical-Case-Reports/Video1.webm
Video 1: Parasternal long axis view shows severe concentric increased wall thickness with left ventricle dysfunction.
https://www.heraldopenaccess.us/fulltext/Clinical-Studies-&-Medical-Case-Reports/Video2.webm
Video 2: Parasternal short axis view shows increased dilatation and concentric wall thickness of left ventricle with reduced contractility
https://www.heraldopenaccess.us/fulltext/Clinical-Studies-&-Medical-Case-Reports/Video3.webm
Video 3: ModifiedApical 4-chamber view shows severe concentric biventricular hypertrophy with severe left ventricle dysfunction
The patient was initially felt to have sarcomeric hypertrophic cardiomyopathy (HCM) presenting in the late dilative phase with heart failure. However, electrical abnormalities such as pre-excitation are infrequent in sarcomeric HCM and raised the possibility of a HCM phenocopy.
Cardiac MRI findings of biventricular hypertrophy with extensive late gadolinium enhancement but sparing the mid septum further raised the suspicion of infiltrative rather than HCM (Figure 2).
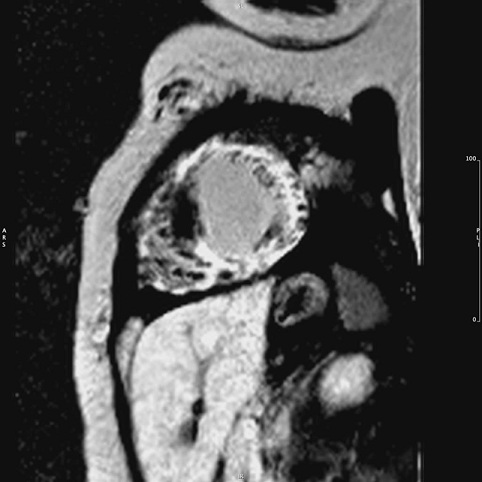 Figure 2: Cardiac MRI showing biventricular hypertrophy with extensive late gadolinium enhancement but sparing the mid septum.
Figure 2: Cardiac MRI showing biventricular hypertrophy with extensive late gadolinium enhancement but sparing the mid septum.
Moreover, the patient also had signs and symptoms of neurologic and retinal involvement, therefore a syndromic pattern explaining all the elements seemed to be more likely.
Genetic tests reported a LAMP2 missense mutation, c.815T>G (p.Leu272Arg).
Management And Follow-Up
Since the clinical condition became critical, the patient was given mechanical support using biventricular ventricular assist devices (BIVAD) (60 mL bilateral Berlin Hearts) and, a week later, the patient underwent an orthotopic heart transplant.
A cardiac muscle specimen from the explanted heart showed extensive fibrosis and cardiomyocytes vacuolization (Figure 3). Immunohistochemical investigation revealed LAMP2 protein staining in the minority of cardiac myocytes, consistent with the p.Leu272Arg loss of function mutation resulting in absence of LAMP2 protein expression (Figure 4). The heterogenous expression of LAMP-2 was speculated to be consistent with variable x-inactivation in cardiomyocytes, a phenomenon also called Lyonization. In her 12-month follow-up visit, the patient was in good conditions and without progression of systemic symptoms.
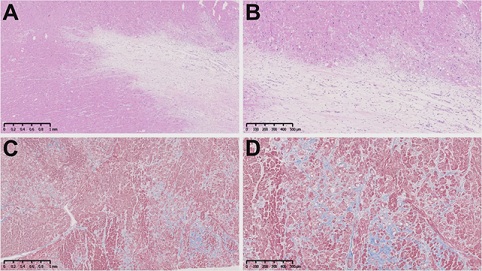 Figure 3: Cardiac muscle specimen from the patient. A and B): hematoxylin and eosin stains showing extensive fibrosis and cardiomyocytes vacuolization (Scale bars: 1mm and 500mm); C and D): trichrome stain showing in blue collagen deposition (Scale bars: 1mm and 500mm).
Figure 3: Cardiac muscle specimen from the patient. A and B): hematoxylin and eosin stains showing extensive fibrosis and cardiomyocytes vacuolization (Scale bars: 1mm and 500mm); C and D): trichrome stain showing in blue collagen deposition (Scale bars: 1mm and 500mm).
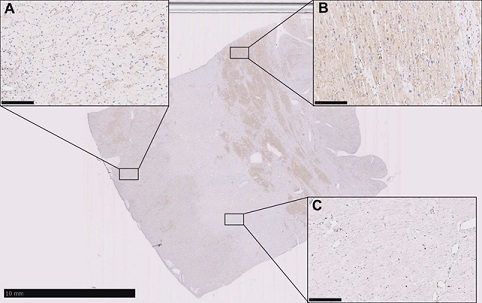 Figure 4: Immunohistochemical analysis on a cardiac muscle specimen showing areas without LAMP-2 expression A): areas with LAMP-2 expression; B): and areas with extensive fibrosis; C): (Scale bars: 10 mm and 250 mm).
Figure 4: Immunohistochemical analysis on a cardiac muscle specimen showing areas without LAMP-2 expression A): areas with LAMP-2 expression; B): and areas with extensive fibrosis; C): (Scale bars: 10 mm and 250 mm).
Discussion
HCM is a heterogeneous group of inherited myocardial diseases which can be divided in sarcomeric HCM, caused by mutations in the gene encoding for cardiac sarcomere, and non-sarcomeric HCM, including Danon Disease (DD).
DD is a rare, X-linked disease which main feature is a vacuolar myopathy caused by mutations in the Lysosomal-Associated Membrane Protein 2 (LAMP2) gene. The protein encoded by this gene has a pivotal role in cellular autophagy, and mutations causing absence or reduced production of the protein result in intracytoplasmatic vacuoles including autophagic material [1,2]. As a consequence, cardiac and skeletal muscles are involved in the clinical picture, but neurodegenerative disorders and pigmentary retinopathy are common as well.
Compared to the sarcomeric HCM disorders, ismore devastating with progressive rapid decline requiring heart transplant at a very young age. Clinical presentation in DD ranges from an isolated heart involvement, often misdiagnosed as sarcomeric HCM, to a multisystemic disorder including cognitive impairment, retinopathy and skeletal myopathy depending on gender, type and location of the mutation in the LAMP-2 gene [3]. This case is illustrative of a fulminant presentation of Danon disease in a young female. As an X-linked syndrome, there is a misconception that DD affects severely only males, and this disease may therefore be overlooked in females [4,5]. However, symptoms can be just as evident in both affected sexes [6]. The reason why some female patients present from the beginning with such severe symptoms and signs and DD’s progression is so rapid remains unclear. Recent basic scientific studies using pluripotent stem cells from monozygotic female twins affected by DD with different severity in clinical manifestations, suggest that such differences may be justified by the lyonization phenomenon by which one of the copies of X-chromosome is inactivated [7]. In this case we noted a heterogenous expression of LAMP2 within the myocardium which is consistent with lyonization. We speculate that the reason this patient had such severe disease is that in her case the majority of X chromosomes were expressing the loss of function mutation. Further research is required to understand the process in which lyonization occurs.
Conclusion
Herein we described a young female who presented with Heart Failure (HF) and severe hypertrophic cardiomyopathy, initially felt to have sarcomeric hypertrophic cardiomyopathy who was ultimately diagnosed with DD. The therapies typically used for HCM and DCM (e.g., myomectomy, neurohormonal therapy) may not be affective in DD, and clinical trials evaluating gene therapy for the disease are ongoing (need references). This case highlights the importance of awareness of inherited rare diseases when evaluating patients with multiple symptoms, and particularly for DD in patients, regardless of sex. More research is required to understand the pathobiology of x linked cardiomyopathies in females.
Learning Objectives
- • To increase awareness about Danon Disease in both males and young females which are usually expected to be less severely affected
- • To be able to make differential diagnosis of sarcomeric HCM and non-sarcomeric HCM with multimodality imaging and histological examination
Conflict of Interest Statement
Eric Adler has an equity interest in Rocket Pharmaceuticals (New York, NY, U.S.A.) and is a co-founder of GenStem Pharmaceuticals (San Diego, CA, U.S.A.).
Michela Brambatti was a previous consultant for Rocket Pharmaceuticals
Matthew Taylor, Ana Manso are consultants for Rocket Pharmaceuticals (New York, NY, U.S.A.).
Marzia Rigolli, Alessandro Maolo, Boss Le and Victor Escobedo have no conflicts of interest or financial ties to disclose.
References
- Nishino I, Fu J, Tanji K, Shimojo S, Koori T, et al. (2000) Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 406: 906-910.
- Eskelinen E-L (2005) Maturation of autophagic vacuoles in Mammalian cells. Autophagy 1: 1-10.
- D’souza RS, Levandowski C, Slavov D, Graw SL, Allen LA, et al. (2014) Danon Disease: Clinical Features, Evaluation, and Management. Circ Heart Fail 7: 843-
- Charron P, Villard E, Sebillon P, Maisonobe T, Duboscq-Bidot L, et al. (2004) Danon’s disease as a cause of hypertrophic cardiomyopathy: a systematic survey. Heart 90: 842-846.
- Yang Z, McMahon CJ, Smith LR, Bersola J, Adesina AM, et al. (2005) Danon disease as an underrecognized cause of hypertrophic cardiomyopathy in children. Circulation 112: 1612-1617.
- Brambatti M, Caspi O, Maolo A, Koshi E, Greenberg B, et al. (2019) Danon Disease: Gender Differences in Presentation and Outcomes. Int J Cardiol 286: 92-98.
- Yoshinda S, Nakanishi C, Okada H, Mori M, Yokawa J, et al. (2018) Characteristics of inducedpluripotentstemcells from clinicallydivergentfemalemonozygotic twins with Danon disease. J Mol Cell Cardiol 114: 234-242.
Citation: Maolo A, Le B, Brambatti M, Escobedo V, Manso AM, et al. (2021) Fulminant Danon Disease in a Young Female Patient with a novel LAMP2 Mutation. J Clin Stud Med Case Rep 8: 0119.
Copyright: © 2020 Alessandro Maolo, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.