Histopathological Characterization of a Rare Case of Soft Tissue Malignant Myoepithelioma: A Diagnostic Challenge
*Corresponding Author(s):
Hiroshi SonobeDepartment Of Diagnostic Pathology, National Hospital Organization (NHO) Fukuyama Medical Center, Hiroshima, Japan
Tel:+81-090-7147-9897,
Email:hnsonobe@gmail.com
Abstract
Background: Soft tissue myoepitheliomas are rare and exhibit a wide spectrum of benignity and low- to high-grade malignancy with histopathological heterogeneity, including cell morphology, nuclear atypia, proliferation patterns, and background matrices, often making pathological diagnosis very challenging. Although recent molecular and genetic studies of the genetic abnormalities, particularly unique gene fusions, have determined associations with the clinicopathological features, they have not been sufficiently elucidated.
Case Presentation: We present a rare case of malignant myoepithelioma that developed in the soft tissue of the groin in an elderly man, along with its macroscopic, histological, immunohistochemical and fluorescence in situ hybridization (FISH) findings. The tumor invaded the adjacent fatty tissue, but no lymph node metastasis was observed locally. Histologically, the tumor cells exhibited severe nuclear atypia, pathological nuclear mitosis, myxoid background, and rhabdoid cells. INI1/SMARCB1 nuclear loss and frequent Ki-67 and p53 positivity indicated a malignancy. Hence, we considered soft tissue malignancies with similar or overlapping histology, such as extra skeletal myxoid chondrosarcoma, proximal epithelioid sarcoma, extrarenal rhabdoid tumor, myxoepithelioid tumor with chordoid features, and malignant myoepithelioma for the differential diagnosis. We used a panel of antibodies, including epithelial membrane antigen (EMA) and cytokeratin’s (CKs) as epithelial markers; vimentin, CD34, desmin and brachyury as mesenchymal markers; synaptophysin, CD56 and insulinoma-associated protein 1 as neuroendocrine markers; and p63, S-100, CD10, alpha-smooth muscle actin, caldesmon and calponin as myoepithelial markers. The tumor was positive for EMA, CK, vimentin, S-100 and CD10, but not for the other markers, and a pathological diagnosis of malignant myoepithelioma was established. The Ewing sarcoma RNA-binding protein 1 (EWSR1) and fused in sarcoma (FUS)-related fusion genes, which have been detected in half of the soft tissue myoepithelioma cases, were not detected upon split FISH. Therefore, a certain fusion gene that is distinct from the EWSR1 or FUS-related genes could be present in the present tumor.
Conclusion: Malignant myoepithelioma diagnosis is very challenging owing to its rarity and clinicopathological diversity. Thus, the possibility of malignant myoepithelioma should always be considered when encountering a soft tissue malignancy that is pathologically questionable, such as the present tumor, which served as a valuable and instructive case.
Keywords
Differential diagnoses; Fusion gene; Immunohistochemistry; Malignant myoepithelioma; Soft tissue
Introduction
Myoepitheliomas are uncommon tumors that develop in the salivary glands, mammary glands, lacrimal glands, sweat glands, and various glands of the respiratory tract, where myoepithelial cells exist in the ducts and acini. However, myoepitheliomas also arise in the soft tissues, bones and various organs without myoepithelial cells [1-11]. Myoepitheliomas occurring in the soft tissues are extremely rare. Soft tissue myoepitheliomas occur clinically in a wide range of ages, from infants to the elderly, and they exhibit a wide spectrum of benign, low malignant, and high malignant potentials. Furthermore, histologically, the tumor cells are oval to spindle-shaped, with nuclear atypia ranging from mild to severe. Therefore, pathological diagnosis is often challenging owing to the diverse clinicopathological characteristics [9,10].
Recently, we encountered an exceedingly challenging case of soft tissue malignancy that developed in the groin of an elderly male patient. For the pathological diagnosis of the present tumor, we considered tumors, such as extra-skeletal myxoid chondrosarcoma (EMC) [12], proximal epithelioid sarcoma (ES) [13], extrarenal rhabdoid tumor (RT) [14,15], myxoepithelioid tumor of chordoid features (METC) [16], and malignant myoepithelioma [9], that mimic the clinical and/or histopathological characteristics. Thus, we were able to diagnose this challenging tumor as a malignant myoepithelioma, using a panel of antibodies as epithelial, mesenchymal and neuroendocrine markers. Although we checked for a EWSR1 or FUS related fusion gene in the present tumor using split fluorescence in situ hybridization (FISH) [17], no fusion gene was detected. Therefore, another type of fusion gene could be present in this case. In soft tissue myoepitheliomas, although associations between genetically and molecular abnormalities, including fusion genes, and clinicopathological findings are being determined, they have not been sufficiently elucidated yet. Further studies and the accumulation of the data are required.
Case Presentation
A male patient in his mid-sixties has visited the urology department of our hospital for a painless mass in his left groin for two months. His family and medical history were unremarkable. A computed tomography (CT) scan revealed a deep soft tissue lesion with a maximum diameter of 37 mm (Figure 1), showing an unclear border with the spermatic cord. The lesion was weakly mobile upon ultrasound examination. The clinical diagnosis of spermatic cord tumor was suspected based on these findings. However, surgery revealed that the lesion was unrelated to the inguinal canal, and no regional or distant metastases were observed. Before surgery, the titers of C-reactive protein (CRP) and tumor markers including carcinoembryonic antigen (CEA), alpha-fetoprotein (AFP), and carbohydrate antigen 19-9 (CA19-9) were within normal ranges. Fifteen months after surgery, no signs of recurrence, lymph node metastasis or distant metastasis were observed, and follow-up is being continued currently.
Macroscopically, the cut-surfaces of the lesion were solid, grayish-white and relatively well-defined with lobulated patterns (Figure 2). Histologically, polygonal to short spindle-shaped tumor cells harbored highly atypical oval nuclei containing distinct nucleoli, and displayed irregular cord-like, reticular and nest-like patterns by tumor cells against a background of myxoid or fibrous stroma of varying degrees in the lesion. Rhabdoid cells were mixed in various proportions. Nuclear mitotic figures including pathological mitotic figures were sporadically observed. No hemorrhagic or necrotic foci were observed (Figures 3a-d). A solid and densely proliferating area of tumor cells was observed only in a small portion at the periphery of the lesion. In addition, the tumor cells partly invaded the fine fibrous capsule and adjacent fatty tissue (Figures 3e,f). Immunohistochemically, the tumor cells showed INI1/hSNF5/SWI/SNF related, matrix associated, actin-dependent regulator of chromatin, subfamily B, member 1 (INI1/SMARCB1) nuclear loss, a Ki-67 index of 40 and a p53 positive rate of 20%, suggesting malignancy (Figures 3g-i).
Thus, EMC, proximal ES, extrarenal RT, METC and malignant myoepithelioma were considered for the differential diagnosis of the present tumor, We incorporated a panel of antibodies including epithelial membrane antigen (EMA), cytokeratin (CK)-AE1/AE3 (AE1/AE3) and CK-CK5/CK6 (CK5/CK6) as epithelial markers; vimentin, CD34, desmin and brachyury as mesenchymal markers; synaptophysin, CD56 and insulinoma-associated protein 1 (INSM1) as neuroendocrine markers; p63, S-100 protein (S-100), CD34, alpha-smooth muscle actin (SMA), calponin and h-caldesmon (caldesmon) as myoepithelial markers; and estrogen receptor (ER) as a hormonal marker. The tumor cells were practically positive for EMA, frequently positive for ER and sporadically positive for AE1/AE3, vimentin, S-100 and CD10, respectively, but not for p63, CK5/CK6, CD34, desmin, SMA, INSM1, synaptophysin, CD56 or brachyury (Figures 4a-i). Thus, the present tumor was pathologically diagnosed as malignant myoepithelioma. To detect a fusion gene in the tumor, Ewing sarcoma RNA-binding protein 1 (EWSR1) and fused in sarcoma (FUS) were examined using the split fluorescence in situ hybridization (FISH) method; however, no split signals were observed (Figures 5a and b).
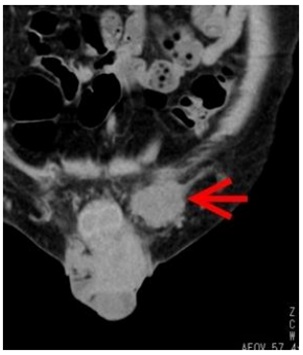 Figure 1: Computed tomography scan: A tumor, 37mm in maximum size, in the left inguinal deep soft tissue (red arrow).
Figure 1: Computed tomography scan: A tumor, 37mm in maximum size, in the left inguinal deep soft tissue (red arrow).
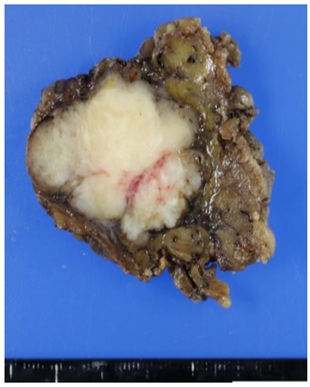 Figure 2: Macroscopic findings: A cut surface of grayish-white and solid tumor with a lobulated pattern and relatively clear borders.
Figure 2: Macroscopic findings: A cut surface of grayish-white and solid tumor with a lobulated pattern and relatively clear borders.
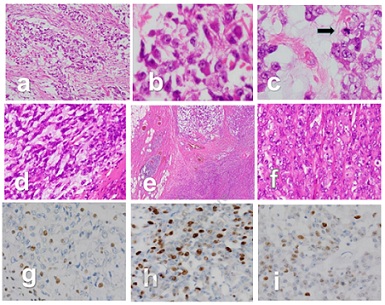 Figure 3: Microscopical and immunohistochemical findings: (a) Tumor cells proliferating in varying sized nests and with a background of fibrous and myxoid matrices 10x. (b) A cell nest consisting of polygonal to short spindle-shaped highly atypical cells with rhabdoid cells 40x. (c) A section showing a nuclear mitotic figure (black arrow) in a myxoid matrix 40x. (d) A section showing spindle-tumor cell proliferation with a myxoid background 20x. (e) A section of tumor cells invading the surrounding adipose tissue 4x. (f) A section showing solid proliferation of polygonal shaped tumor cells 20x. Immunohistochemically, the tumor cells showing (g) INI1 nuclear loss, and (h) Ki-67 index 40 and (i) 20% p53 positivity.
Figure 3: Microscopical and immunohistochemical findings: (a) Tumor cells proliferating in varying sized nests and with a background of fibrous and myxoid matrices 10x. (b) A cell nest consisting of polygonal to short spindle-shaped highly atypical cells with rhabdoid cells 40x. (c) A section showing a nuclear mitotic figure (black arrow) in a myxoid matrix 40x. (d) A section showing spindle-tumor cell proliferation with a myxoid background 20x. (e) A section of tumor cells invading the surrounding adipose tissue 4x. (f) A section showing solid proliferation of polygonal shaped tumor cells 20x. Immunohistochemically, the tumor cells showing (g) INI1 nuclear loss, and (h) Ki-67 index 40 and (i) 20% p53 positivity.
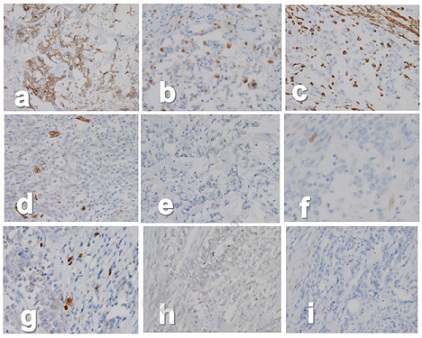
Figure 4: Immunohistochemical findings: The tumor cells being frequently positive for epithelial membrane antigen (EMA) (a), often positive for cytokeratin-AE1/AE3 (AE1/AE3) (b), vimentin (c), negative for CD34 (d) and insulinoma-associated protein 1 (INSM1) (e), occasionally positive for CD10 (f) and protein S-100 (S-100) (g), and negative for brachyury (h) and desmin (i).
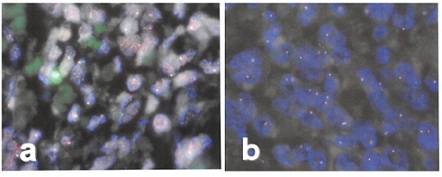
Figure 5: Split fluorescence in situ hybridization (FISH) findings: (a) No detection of Ewing sarcoma RNA-binding protein 1 (EWSR1) (a) or (b) fused in sarcoma (FUS) translocation. Tumor cells showing two fusion signals, one indicated orange color and the other in green color.
Discussion
As already mentioned, the tumor in the present case developed in the deep soft tissue of the groin. Epithelioid to short-spindle cells with severe nuclear atypia and rhabdoid cells proliferated in various patterns on the background of the myxoid stroma. The tumor cells demonstrated INI1/SMARCB1 nuclear loss and a high incidence of Ki-67 and p53 positivity. Thus, we considered the following soft tissue malignancies for the differential diagnosis: EMC [12], proximal ES [13], extrarenal RT [14,15], METC [16] and malignant myoepithelioma [9,10]. However, all the tumors are rare and exhibit similar or overlapping histological features; furthermore, their origin or line of differentiation is unknown. Therefore, their pathological diagnosis is often confusing or challenging.
Considering the clinical and pathological characteristics as well as the unique fusion genes in the tumors listed above, EMC typically reveals mild to moderate nuclear atypia and positivity for vimentin, S100, EMA and neuroendocrine markers such as synaptophysin, CD56 and INSM1 [12,18]. The rare solid and cellular variant of high-grade malignancy densely consists of atypical epithelioid cells and pleomorphic or rhabdoid cells that show INI1/SMARCB1 nuclear loss [19]. Nuclear receptor subfamily 4 group A member 3 (NR4A3)-related fusion genes lacking EWSR1 as a fusion partner have also been detected [20]. Proximal ES is characterized by a solid proliferation of highly atypical epithelioid cells with a number of rhabdoid cells [13] and seldom harbors a remarkable myxoid background [21]. Immunohistochemically, the tumor cells are diffusely and strongly positive for vimentin, CK and CD34, and moreover, INI1/SMARCB1 nuclear loss is observed in most cases [22]. No unique fusion genes have been described for this tumor. Extrarenal RT shows very similar histological features to proximal ES, but is more prevalent in children and the tumor cells are negative for CD34. Yoshida, et al. [23] in 2015 advocated myoepitheliod-like tumors of the vulvar region (METVR) as a new concept. This low-grade malignant tumor develops exclusively in women, and epithelial-like to spindle-shaped tumor cells with moderate nuclear atypia proliferate in cord-like, reticular and nest-like patterns against a background of abundant myxoid stroma. The tumor cells are positive for ER and SMA, and for CK in a minority, but not for S-100, GFAP and CD34 [23,24]. In 2021, Kinoshita, et al. [16] proposed the concept of myxoepithelioid tumor with chordoid features (METC) that encompasses METVR. It exhibits the same histological features as those of METVR and develops in both women and men. The tumor cells are positive for brachyury in METC. METVR and METC show rhabdoid cells and INI1/SMARCB1 nuclear loss upon immunostaining. No specific fusion gene is identified. METC and METVR recently proposed as a new entity are still not described in the World Health Organization (WHO) classification of often tissue and bone tumors (5th edition) [25].
Since INI1/SMARCB1 nuclear loss of tumor cells and the appearance of rhabdoid cells may exist in the above-mentioned soft tissue tumors, these findings are not necessarily useful for differentiation among them. Moreover, EMC is positive for neuroendocrine markers, whereas the present tumor was negative. Proximal ES frequently exhibits positivity for vimentin, CK, EMA and CD34, and it seldom harbors a myxoid background. Extrarenal RT displays similar histological findings and is frequently positive for vimentin and CK but not for CD34 [13], and develops exclusively in children [14,15]. The present tumor occurred in an elderly patient and had an apparent myxoid background [20]. Immunohistochemically, it showed only sporadic positivity for vimentin and CK, but not for CD34. Based on these findings, the present tumor does not correspond to proximal ES or extrarenal RT. Although METC appears histologically very similar to the present tumor, METC typically shows weaker nuclear atypia, and positivity for brachyury is essential for its diagnosis [16], while the present tumor showed severe nuclear atypia and negativity for brachyury. Therefore, the present tumor did not correspond to METC.
Myoepitheliomas present a broad clinicopathological spectrum ranging from benign and low-grade to high-grade malignancy, thereby making the diagnosis challenging in the cases being consider. Histologically, polygonal to spindle shaped tumor cells grow in sheet, nest and cord-like patterns, often in myxoid or fibrous to hyaline backgrounds. Tumor cells are positive for myoepithelial markers such as CD10, p63, calponin, caldesmon, GFAP and S100, as well as EMA, CKs and vimentin. The appearance of rhabdoid cells and INI1 nuclear loss of tumor cells have been found in a subset. The histological and immunohistochemical features as described above vary, reflecting the clinical variability between benign to malignant myoepitheliomas. The present tumor was positive for not only EMA, AE1/AE3 and vimentin but also CD10 and S100 as myoepithelial markers. Moreover, Ki67 and p53 were positively detected at high levels, indicating malignancy. Thus, in spite of the complicated case, the pathological diagnosis of the present tumor as a malignant myoepithelioma was finally established.
In recent years, molecular and genetic studies have been conducted on various soft tissue tumors, and tumor-related genetic abnormalities, especially unique fusion genes, have been identified [26-29]. These findings must strengthen or complement a conventional pathological diagnosis based on histological and immunohistochemical findings, anticipating further accumulation of the genetic and molecular findings. Concerning benign to malignant myoepitheliomas, EWSR1- and FUS-related fusion genes with various partner genes have been detected [30-33]. In 2020, Suurmeijer, et al. [17] reported that fusion genes involving EWSR1 and FUS were detected in 49 of 66 cases of benign to malignant myoepitheliomas. Among the 49 cases, EWSR1 was detected in 37, of which the partner genes were POU5F1, PBX3, PBX1, ZNF444, KLF15, and KLF17, whereas FUS was detected in 12 cases with KLF17 or POU5F1 as the partner gene. Of the 66 cases, 17 were malignant, of which 11 had EWSR1-POU5F1 and three possessed EWSR1-ZNF44. Three patients with FUS-KLF17 demonstrated myxoid backgrounds. The cases of EWSR1-PBX1/3 and FUS-PBX1/3 were benign with a fibrotic background. However, no fusion genes were detected in the remaining 17 cases, and therefore, other fusion genes unrelated to EWSR1 and FUS should be present. Actually, benign to malignant soft tissue myoepitheliomas with fusion genes unrelated to EWSR1 and FUS, such as ASCC2-GGNBP2, IRF2BP2-CDX2 and SRF-E2F1, have been reported previously [34-36]. In the present malignant myoepithelioma, EWSR1 and FUS were examined using split FISH; however, no fusion genes were identified.
Although myoepitheliomas arising from different sites exhibit similar histological features, salivary myoepitheliomas usually harbor fusion genes involving PLAG1 [37-40], which are distinct from those of the soft tissue and cutaneous myoepitheliomas involving EWSR1 and FUS [17,41-42]. However, soft tissue myoepitheliomas with fusion genes related to PLAG1 have also been reported [39,43,44]. In addition, cutaneous syncytial myoepitheliomas were reportedly associated with fusion genes related to EWSR1 [41], whereas cutaneous myoepitheliomas with ductal differentiation were associated with PLAG1 related fusion genes [45]. Namely, some overlaps in fusion gene types exists among salivary, cutaneous and soft tissue myoepitheliomas. Hence, the fusion genes in myoepitheliomas have not been fully elucidated. To clarify the relationship between the clinicopathological findings of soft tissue myoepitheliomas and fusion genes in detail, further accumulation of cases of myoepitheliomas arising not only from the soft tissue but also other sites is warranted.
Conclusion
In conclusion, malignant myoepitheliomas of the soft tissue are often challenging to diagnose by histopathological characterization owing to their rarity and diversity in cell morphology, nuclear atypia, proliferation, and background matrices. The present malignant soft tissue myoepithelioma exhibited similar histomorphology to that of EMC, proximal ES, extrarenal RT, and METC. In the present case, a panel of antibodies composed of various makers, including myoepithelial markers, was considered a powerful weapon for establishing the pathological diagnosis. Moreover, the possibility of malignant myoepithelioma should always be considered when encountering a soft tissue malignancy that is pathologically questionable, such as the present tumor, which served as a valuable and instructive case.
List of Abbreviations
FISH: Fluorescence in situ hybridization; EMC: Extra skeletal myxoid chondrosarcoma; ES: Epithelioid sarcoma; RT: Rhabdoid tumor; METC: Myxoepithelioid tumor of choroid features; CT: Computed tomography; CRP: C-reactive protein; CEA: Carcinoembryonic antigen; AFP: Alpha-fetoprotein; CA19-9: Carbohydrate antigen 19-9; EMA: Epithelial membrane antigen; CK: Cytokeratin; AE1/AE3: Cytokeratin-AE1/AE3; INSMI: insulinoma-associated protein 1; S-100: S-100 protein; ER: Estrogen receptor; SMA: Alpha-smooth muscle actin; Caldesmon: h-caldesmon; EWSR1: Ewing sarcoma RNA-binding protein 1; FUS: fused in sarcoma; GAFP: Glial acidic fibrillary protein; NR4A3: Nuclear receptor subfamily 4 group A member 3; METVR: Myoepitheliod-like tumors of the vulvar region; METC: Myxoepithelioid tumor with choroid features.
Declarations
Source of support
None.
Ethics approval and consent to participate
All the procedures including the handling of personal information in this case report were conducted in accordance with the ethical standards of the Helsinki Declaration of 1964 and its later versions by the responsible committee at the NHO Fukuyama Medical Center.
Consent for publication
Written informed consent was obtained from the patient for publication of this article.
Availability of data and materials
All relevant data are within the paper and it’s supporting Information files.
Competing interests
The authors declare that they have no competing interests.
Funding
None.
Authors' contributions
Hiroshi Sonobe was concerned with the conceptualization of the study, study design, data acquisition, data analysis, data interpretation, drafting the manuscript, critical revision of the manuscript for important intellectual content, and final approval of the version to be published.
Hiroshi Sonobe, Rika Omote, Hiroyuki Yanai and Hidetaka Yamamoto discussed the pathological characterization of the present soft tissue tumor and subsequently, arrived at the diagnosis of malignant myoepithelioma.
Kazuma Yukihiro and Hiroshi Masumoto performed the clinical diagnosis based on the patient’s history, symptoms and images, and conducted the surgery and follow-up.
Acknowledgement
We thank all the staffs of the Department of Diagnostic Pathology, NHO Fukuyama Medical Center, for their support in this case, and Dr. Izumi Kinoshita and Prof. Yoshinao Oda, Department of Anatomic Pathology, Pathological Sciences, Graduate School of Medical Sciences. Kyushu University for appropriate suggestions about the pathological diagnosis in this case. We also thank Editage for proofreading this manuscript.
References
- Telugu RB, Gaikwad P, Baitule A, Michael RC, Mani T, et al. (2021) Myoepithelial tumors of salivary gland: a clinicopathologic and immunohistochemical study of 15 patients with MIB-1 correlation. Head Neck Pathol; 15: 479-490.
- Mok Y, Agaimy A, Wang S, Kuick CH, Chang KT, et al. (2018) High-grade myoepithelial carcinoma can show histologically undifferentiated/anaplastic featu3031res. Ann Diagn Pathol. 37: 20-24.
- Huin M, Body G, Arbion F, Ouldamer L (2022) Malignant breast myoepithelioma: a systematic review. Hum Reprod; 51: 102481
- Grossniklaus HE, Wojno TH, Wilson MW, Someren AO (1997) Myoepithelioma of the lacrimal gland. Arch Ophthalmol; 115: 1588-1590.
- Mentzel T, Requena L, Kaddu S, Soares de Aleida LM, Sangueza OP, et al. (2003) Cutaneous myoepithelial neoplasms: clinicopathologic and immunohistochemical study of 20 cases suggesting a continuous spectrum ranging from benign mixed tumor of the skin to cutaneous myoepithelioma and myoepithelial carcinoma. J Cutan Pathol. 30: 294-302.
- Hornick JL, Fletcher CD (2004) Cutaneous myoepithelioma: a clinicopathologic and immunohistochemical study of 14 cases. Hum Pathol. 35: 14-24.
- Gaio E, Perasole A, Bagatella F (2009) Bilateral myoepithelioma of the nasopharynx: a case report. Auris Nasus Larynx. 36: 496-500.
- Chand M, Mann JM, Sabayev V, Luo JJ, Cohen PR, et al. (2011) Endotracheal myoepithelioma. Chest. 140: 242-244.
- Hornick JL, Fletcher CDM (2003) Myoepithelial tumors of soft tissue: a clinicopathologic and immunohistochemical study of 101 cases with evaluation of prognostic parameters. Am J Surg Pathol. 27: 1183-1196.
- Verma A, Rekhi B (2017) Myoepithelial tumor of soft tissue and bone: a current perspective. Histol Histopathol. 32: 861-877.
- Song W, Flucke U, Suurmeijer AJH (2017) Myoepithelial tumors of bone. Surg Pathol Clin. 10: 657-674.
- Goh YW, Spagnolo DV, Platten M, Caterina P, Fisher C, et al. (2001) Extraskeletal myxoid chondrosarcoma: a light microscopic, immunohistochemical, ultrastructural and immuno-ultrastructural study indicating neuroendocrine differentiation. Histopathology. 39: 514-524.
- Guillou L, Wadden C, Coindre JM, Krausz T, Fletcher CD (1997) "Proximal-type" epithelioid sarcoma, a distinctive aggressive neoplasm showing rhabdoid features. Clinicopathologic, immunohistochemical, and ultrastructural study of a series. Am J Surg Pathol. 21: 130-146.
- Fanburg-Smith JC, Hengge M, Hengge UR, Smith JS Jr, Miettinen M (1998) Extrarenal rhabdoid tumors of soft tissue: a clinicopathologic and immunohistochemical study of 18 cases. Ann Diagn Pathol. 2: 351-362.
- Oda Y, Tsuneyoshi M (2006) Extrarenal rhabdoid tumors of soft tissue: clinicopathological and molecular genetic review and distinction from other soft-tissue sarcomas with rhabdoid features. Pathol Int. 56: 287-295.
- Kinoshita I, Kohashi K, Yamamoto H, Yamada Y, Inoue T, et al. (2021) Myxoepithelioid tumour with chordoid features: a clinicopathological, immunohistochemical and genetic study of 14 cases of SMARCB1/INI1-deficient soft-tissue neoplasm. Histopathology.79: 629-641.
- Suurmeijer AJH, Dickson BC, Swanson D, Zhang L, Sung YS, et al. (2020) A morphologic and molecular reappraisal of myoepithelial tumors of soft tissue, bone, and viscera with EWSR1 and FUS gene rearrangements. Genes Chromosomes Cancer. 59: 348-356.
- Yoshida A, Makise N, Wakai S, Kawai A, Hiraoka N (2018) INSM1 expression and its diagnostic significance in extraskeletal myxoid chondrosarcoma. Mod Pathol. 31: 744-752.
- Hollmann TJ, Hornick JL (2011) INI1-deficient tumors: Diagnostic features and molecular genetics. Am J Surg Pathol. 35: e47-e63.
- Flucke U, Tops BB, Verdijk MA, van Cleef PJ, van Zwam PH, et al. (2012) NR4A3 rearrangement reliably distinguishes between the clinicopathologically overlapping entities myoepithelial carcinoma of soft tissue and cellular extraskeletal myxoid chondrosarcoma. Virchows Arch. 460: 621-628.
- Flucke U, Hulsebos TJM, van Krieken JHJM, Mentzel T (2010) Myxoid epithelioid sarcoma: a diagnostic challenge. A report on six cases. Histopathology. 57: 753-759.
- Hornick JL, Dal Cin P, Fletcher CD (2009) Loss of INI1 expression is characteristic of both conventional and proximal type epitheliod sarcoma. Am J of Surg Pathol. 33: 542-550.
- Yoshida A, Yoshida H, Yoshida M, Mori T, Kobayashi E, et al. (2015) Myoepithelioma-like tumors of the vulvar region: a distinctive group of SMARCB1-deficient neoplasms. Am J Surg Pathol. 39: 1102-1113.
- Xu Y, Gao H, Gao JL (2020) Myoepithelioma-like tumor of the vulvar region: a case report in China and review of the literature. Diagn Pathol. 8: 15:3.
- Fletcher CDM, Baldini EH, Blay JY, Gronchi A, Lazar AJ, et al. (2020) Soft tissue tumours: Introduction. In: The WHO Classification of Tumours Editorial Board, editor. WHO Classification of Tumours. Soft Tissue and Bone Tumours. 5th ed. IARC Press; Lyon, France: 2020: 6-12.
- Fisher C (2014) The diversity of soft tissue tumours with EWSR1 gene rearrangements: a review. Histopathology. 64: 134-150.
- Flucke U, van Noesel MM, Siozopoulou V, Creytens D, Tops BBJ, et al. (2021) EWSR1-The most common rearranged gene in soft tissue lesions, which also occurs in different bone lesions: an updated review. Diagnostics (Basel). 11: 1093.
- Choi JH, Ro JY (2023) The recent advances in molecular diagnosis of soft tissue tumors. Int J Mol Sci. 24: 5934.
- Antonescu CR, Zhang L, Chang N-E, Pawel BR, Travis W, et al. (2010) EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. A molecular analysis of sixty-six cases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer. 49: 1114-1124.
- Huang SC, Chen HW, Zhang L, Sung YS, Agaram NP, et al. (2015) Novel FUS-KLF17 and EWSR1-KLF17 fusions in myoepithelial tumors. Genes Chromosomes Cancer. 54: 267-275.
- Leduc C, Zhang L, Öz B, Luo J, Fukuoka J, et al. (2016) Thoracic myoepithelial tumors: a pathologic and molecular study of 8 cases with review of the literature. Am J Surg Pathol. 40: 212-223.
- Komatsu M, Kawamoto T, Kanzawa M, Kawakami Y, Hara H, et al. (2020) A novel EWSR1-VGLL1 gene fusion in a soft tissue malignant myoepithelial tumor. Genes Chromosomes Cancer. 59: 249-254.
- Leckey BD Jr, John I, Reyes-Múgica M, Naous R (2021) EWSR1-ATF1 Fusion in a myoepithelial carcinoma of soft tissue with small round cell morphology: a potential diagnostic pitfall. Pediatr Dev Pathol. 24: 258-263.
- Cyrta J, Rosiene J, Bareja R, Kudman S, Al Zoughbi W, et al. (2022) Whole-genome characterization of myoepithelial carcinomas of the soft tissue. Cold Spring Harb Mol Case Stud. 8: a006227.
- Patton A, Speeckaert A, Zeltman M, Cui X, Oghumu S, et al. (2023) A novel IRF2BP2::CDX2 2Gene fusion in digital intravascular myoepithelioma of soft tissue: an enigma. Genes Chromosomes Cancer. 62: 176-183.
- Urbini M, Astolfi A, Indio V, Tarantino G, Serravalle S, et al. (2017) Identification of SRF-E2F1 fusion transcript in EWSR-negative myoepithelioma of the soft tissue. Oncotarget. 8: 60036-60045.
- Xu B, Katabi N (2021) Myoepithelial Carcinoma. Surg Pathol Clin. 14: 67-73.
- Rupp NJ, Höller S, Brada M, Vital D, Morand GB, et al. (2022) Expanding the clinicopathological spectrum of TGFBR3-PLAG1 rearranged salivary gland neoplasms with myoepithelial differentiation including evidence of high-grade transformation. Genes Chromosomes Cancer. 61: 94-104.
- Dalin MG, Katabi N, Persson M, Lee KW, Makarov V, et al. (2017) Multi-dimensional genomic analysis of myoepithelial carcinoma identifies prevalent oncogenic gene fusions. Nat Commun. 8: 1197.
- Skálová A, Agaimy A, Vanecek T, Banecková M, Laco J, et al. (2021) Molecular profiling of clear cell myoepithelial carcinoma of salivary glands with EWSR1 rearrangement identifies frequent PLAG1 gene fusions but no EWSR1 fusion transcripts. Am J Surg Pathol. 45: 1-13.
- Jo VY, Antonescu CR, Dickson BC, Swanson D, Zhang L, et al. (2019) Cutaneous syncytial myoepithelioma is characterized by recurrent EWSR1-PBX3 fusions. Am J Surg Pathol. 43: 1349-1354.
- Flucke U, Palmedo G, Blankenhorn N, Slootweg PJ, Kutzner H, et al. (2011) EWSR1 gene rearrangement occurs in a subset of cutaneous myoepithelial tumors: a study of 18 cases. 24: 1444-1450.
- Matsuyama A, Hisaoka M, Hashimoto H (2012) PLAG1 expression in mesenchymal tumors: an immunohistochemical study with special emphasis on the pathogenetical distinction between soft tissue myoepithelioma and pleomorphic adenoma of the salivary gland. Pathol Int. 62: 1-7.
- Segawa K, Sugita S, Aoyama T, Takenami T, Asanuma H, et al. (2020) Myoepithelioma of soft tissue and bone, and myoepithelioma-like tumors of the vulvar region: Clinicopathological study of 15 cases by PLAG1 immunohistochemistry. Pathol Int. 70: 965-974.
- Antonescu CR, Zhang L, Shao SY, Mosquera JM, Weinreb I, et al. (2013) Frequent PLAG1 gene rearrangements in skin and soft tissue myoepithelioma with ductal differentiation. Genes Chromosomes Cancer. 52: 675-82.
Citation: Sonobe H, Omote R, Yukihiro K, Masumoto H, Yanai H, et al. (2023) Histopathological Characterization of a Rare Case of Soft Tissue Malignant Myoepithelioma: A Diagnostic Challenge. J Clin Stud Med Case Rep 10:178.
Copyright: © 2023 Hiroshi Sonobe, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.