Journal of Neonatology & Clinical Pediatrics Category: Clinical
Type: Research Article
Hypocomplementemic Urticarial Vasculitis in a Pediatric Patient
*Corresponding Author(s):
Elif DokmeciPediatric Allergy And Clinical Immunology, Yale University School Of Medicine, 333 Cedar Street, PO Box 208064, New Haven, CT 06520-8064, United States
Tel:+1 2037377175,
Fax:+1 2037376035
Email:elif.dokmeci@yale.edu
Received Date: Feb 18, 2015
Accepted Date: Mar 24, 2015
Published Date: Apr 07, 2015
Abstract
Hypocomplementemic urticarial vasculitis syndrome or McDuffie syndrome may go undiagnosed for many years as its initial presentation is similar to allergic urticaria. However, there are key differences in the history as well as clinical course that differentiate this syndrome from its other well-known counterpart. Urticarial vasculitis mainly involves the skin, but might affect other organ systems. If it is associated with low complement levels, it might cause extensive vasculitis and systemic features that most commonly involve the musculoskeletal, pulmonary, renal, and/or gastrointestinal systems. Although urticarial vasculitis is most commonly idiopathic, it can occur in association with autoimmune diseases, drug reactions, infections, or malignancy. This report will highlight these differences, review the pathogenesis of this disease and briefly discuss the associated co-morbidities.
Keywords
Anti-C1q vasculitis; HUVS; Hypocomplementemic urticarial vasculitis
ABBREVIATIONS
C: Complement
HUVS: Hypocomplementemic Urticarial Vasculitis Syndrome
SLE: Systemic Lupus Erythematosus
UV: Urticarial Vasculitis
HUVS: Hypocomplementemic Urticarial Vasculitis Syndrome
SLE: Systemic Lupus Erythematosus
UV: Urticarial Vasculitis
INTRODUCTION
Urticarial Vasculitis (UV) is an immune-complex mediated pathologic condition that is characterized by a variety of cutaneous, systemic, and serologic features [1,2]. Patients present with generalized urticarial eruptions located on the trunk, proximal extremities and face. These lesions are often associated with itching or pain and persist for more than 24 hours. Hyperpigmentation develops after resolution of the urticarial lesions. Angioedema is common and may also be a presenting feature.
Although UV is most commonly idiopathic, it can occur in association with autoimmune diseases, drug reactions, infections, or malignancy. UV likely represents a continuum of disease, ranging from urticaria with minimal vasculitis, to life-or organ-threatening systemic vasculitis with minimal urticaria. Some patients have low complement levels, a feature that is associated with more severe disease and with systemic involvement. The prevalence of UV is 5% to 10% in patients with chronic urticarial [3]. Urticarial vasculitis is very rare in children, with only few pediatric cases reported [4-7]. Patients most commonly present during the fourth decade of life. Most patients are white with a female-male ratio of 8:1 [2,8].
Although UV is most commonly idiopathic, it can occur in association with autoimmune diseases, drug reactions, infections, or malignancy. UV likely represents a continuum of disease, ranging from urticaria with minimal vasculitis, to life-or organ-threatening systemic vasculitis with minimal urticaria. Some patients have low complement levels, a feature that is associated with more severe disease and with systemic involvement. The prevalence of UV is 5% to 10% in patients with chronic urticarial [3]. Urticarial vasculitis is very rare in children, with only few pediatric cases reported [4-7]. Patients most commonly present during the fourth decade of life. Most patients are white with a female-male ratio of 8:1 [2,8].
Urticarial vasculitis has three distinct syndromes
Normocomplementemic urticarial vasculitis: NUV is generally a benign and self-limited subset with unknown etiology. It is also considered a cutaneous leukocytoclastic angitis. The patients in this group are more likely to have mild disease and normal complement levels.
Hypocomplementemic urticarial vasculitis: HUV is an immune complex-mediated small vessel vasculitis characterized by urticaria and hypocomplementemia (low C1q with or without low C3 and C4), and usually associated with circulating anti-C1q autoantibodies. It is divided into two subtypes. The primary or idiopathic formis not associated with systemic disease. The secondary form is due to a chronic inflammatory disease [9]. The secondary form also has overlapping features of Systemic Lupus Erythematosus (SLE) with low Complement (C) levels, autoantibodies and complement deposition at dermal-epidermal junction [10].
HUV syndrome: HUVS is a rare and severe form of UV with systemic and multiorgan involvement. Its etiology is not known. It is characterized by persistent urticarial skin lesions, leucocytoclastic vasculitis, and a variety of systemic manifestations, including severe angioedema, laryngeal edema, ocular inflammation, arthritis, arthralgia, obstructive lung disease, recurrent abdominal pain, and glomerulonephritis [11]. Due to absence of ANA in HUVS, its association with other diseases is still unclear [8].
Some authors have suggested the possible progression of HUVS to SLE, but others consider HUVS as an independent immunological disease from SLE.
According to Davis and Brewer, HUV, SLE-like syndrome, and HUVS, may be a continuum of the same disease, ranging from urticaria with minimal vasculitis to urticarial with marked systemic vasculitis and associated hypocomplementemia [1,12]. On the other hand 20 years of retrospective and prospective observations in patients with NUV, HUV, and HUVS did not support the hypothesis of transition among these UV subtypes [2].
Diagnosis of HUV requires the presence of two major criteria (recurrent urticaria for > 6 months and hypocomplementemia) and at least two minor criteria (leukocytoclastic vasculitis on biopsy, arthralgia and arthritis, ocular inflammation, abdominal pain, glomerulonephritis and positive C1q precipitin test by immunodiffusion with an associated suppressed C1q level) [1]. Urticarial vasculitis is a rare diagnosis. It is challenging to diagnose these cases due to a lack of clarity in the medical literature as there are variety of cutaneous, systemic, and serologic features associated with the disease. It is very rare in children, only a few case reports are present in the literature. Here, we report a 6 year old child with hypocomplementemic urticarial vasculitis who started to have symptoms during her first year of life.
Hypocomplementemic urticarial vasculitis: HUV is an immune complex-mediated small vessel vasculitis characterized by urticaria and hypocomplementemia (low C1q with or without low C3 and C4), and usually associated with circulating anti-C1q autoantibodies. It is divided into two subtypes. The primary or idiopathic formis not associated with systemic disease. The secondary form is due to a chronic inflammatory disease [9]. The secondary form also has overlapping features of Systemic Lupus Erythematosus (SLE) with low Complement (C) levels, autoantibodies and complement deposition at dermal-epidermal junction [10].
HUV syndrome: HUVS is a rare and severe form of UV with systemic and multiorgan involvement. Its etiology is not known. It is characterized by persistent urticarial skin lesions, leucocytoclastic vasculitis, and a variety of systemic manifestations, including severe angioedema, laryngeal edema, ocular inflammation, arthritis, arthralgia, obstructive lung disease, recurrent abdominal pain, and glomerulonephritis [11]. Due to absence of ANA in HUVS, its association with other diseases is still unclear [8].
Some authors have suggested the possible progression of HUVS to SLE, but others consider HUVS as an independent immunological disease from SLE.
According to Davis and Brewer, HUV, SLE-like syndrome, and HUVS, may be a continuum of the same disease, ranging from urticaria with minimal vasculitis to urticarial with marked systemic vasculitis and associated hypocomplementemia [1,12]. On the other hand 20 years of retrospective and prospective observations in patients with NUV, HUV, and HUVS did not support the hypothesis of transition among these UV subtypes [2].
Diagnosis of HUV requires the presence of two major criteria (recurrent urticaria for > 6 months and hypocomplementemia) and at least two minor criteria (leukocytoclastic vasculitis on biopsy, arthralgia and arthritis, ocular inflammation, abdominal pain, glomerulonephritis and positive C1q precipitin test by immunodiffusion with an associated suppressed C1q level) [1]. Urticarial vasculitis is a rare diagnosis. It is challenging to diagnose these cases due to a lack of clarity in the medical literature as there are variety of cutaneous, systemic, and serologic features associated with the disease. It is very rare in children, only a few case reports are present in the literature. Here, we report a 6 year old child with hypocomplementemic urticarial vasculitis who started to have symptoms during her first year of life.
PATIENT DESCRIPTION
A 6 year old female presented at 1 year of age with recurrent episodes of urticarial skin rash. The lesions were dark purple to red macules with wheals associated with a burning and stinging sensation, but no to minimal pruritus (Figure 1). She also complained of elbow and knee pain when the rash appeared. Occasionally, she experienced episodes of abdominal pain which she describes as dull pain that started just before or during episodes. She had mild periorbital edema, but no other associated symptoms. The rash lasted for over 24 hours and was poorly responsive to anti-histamines such as cetirizine or fexofenadine. There were no precipitating factors such as environmental changes, food, animal contact, seasonal variation, or infections. When the wheals resolved, there were residual hyperpigmented, dark red-brown macules. She has had many episodes of urticarial rash in the past and none of them responded to antihistamines. When she was treated with 5-10 days of oral prednisolone, her symptoms slowly cleared including her cutaneous manifestations and joint pains. Her family history was non-contributory. The patient’s past medical history included intermittent asthma controlled with inhaled fluticasone and RSV bronchiolitis.
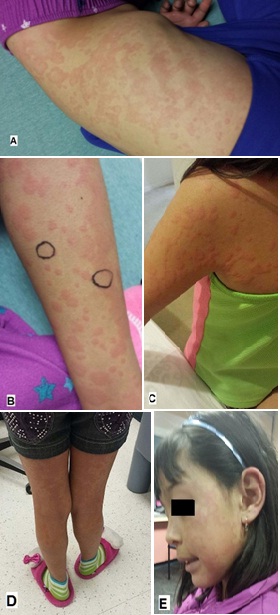
Figure 1: Cutanous manifestation of hypocomplementemic urticarial vasculitis.
A: Shows the dark macules and wheals at presentation on the patient’s abdomen; B and C: Red macules and wheals on the arms and shoulder s of our patient; D and E: Red macules and wheals on the face and legs of our patient. The patient’s rash did not respond to antihistamines and she remained symptomatic for many days. After systemic steroids were started, her symptoms began to resolve and her rash cleared slowly within 3-4 days.
Figure 1: Cutanous manifestation of hypocomplementemic urticarial vasculitis.
A: Shows the dark macules and wheals at presentation on the patient’s abdomen; B and C: Red macules and wheals on the arms and shoulder s of our patient; D and E: Red macules and wheals on the face and legs of our patient. The patient’s rash did not respond to antihistamines and she remained symptomatic for many days. After systemic steroids were started, her symptoms began to resolve and her rash cleared slowly within 3-4 days.
Laboratory studies were significant for a persistently low C4 level (12.1 mg/dL) but normal CH50, C2 and C3 levels. Thyroid function, ANA, ANCA, ESR, CRP, CBC and urinalysis were all normal. She had a mild transaminitis with AST 58 units/L and ALT 405 units/L. Hepatitis B and C serologies were negative. Serum electrophoresis was normal. She also had normal serum Immunoglobulins. Other rheumatologic tests such as Jo-1 antibody, anti-Smith, anti-Scl, anti-dsDNA, cyclic citrinulated peptide, RF, Anti- RNP antibody, SS-A and SS-B precipitins were all normal. These studies and clinical presentation raised suspicion for HUV. Anti-C1q IgG antibody was obtained. It was markedly elevated at 275 units (Normal: 0-19) with positive C1q precipitin assay and low C1q levels.
Based on the presence of highly elevated anti-C1q IgG antibody/ precipitin levels, low C4 and C1q levels and clinical features of urticarial vasculitis including arthralgia and abdominal pains; the patient was diagnosed with hypocomplementemic urticarial vasculitis. She was immediately screened for systemic and organ involvement of the disease. Further work up indicated did not show associated systemic multiorgan disease. She is being closely followed for the development of systemic problems, including urinalysis, eye exams and pulmonary function tests.
PATHOPHYSIOLOGY
Antibodies against C1q is a diagnostic marker for HUVS. However, they are not specific to HUVS since SLE patients also have high levels of antibody. UV lesions are caused by Leukocytoclastic Vasculitis (LCV) in contrast to other forms of urticaria. UV wheals persist for more than 48 hours, but generally between 24 to 72 hours versus common urticaria resolves within 8 to 24 hours. UV lesions are found to cause dysesthesia and less commonly pruritis, which is more associated with common urticaria. UV lesions have a predilection of arising anywhere and they tend to be centripetal, favoring the trunk and proximal extremities. Common urticaria characteristically affects the lower extremities [10,13].The eruptions of HUV are observed on the face, upper extremities, and trunk. They can also present on the palms and soles. The urticarial lesions last for 2 to 4 days then fade without scarring. Angioedema may be found when a vasculitis involves deeper vessels [14].
C1q precipitins (C1q-p also referred to as anti-C1qAb/C1q) are IgG autoantibodies that bind to the Fc portion, collagen-like regions, of the C1 molecule. They form immune complexes and activate the complement system (C3a and C5a) by the classical pathway within and around blood vessels [15]. The activation of this cascade causes mast cell degranulation and generates anaphylatoxins. In addition, it upregulates chemokines and cytokines, which lead to deposition of immune complexes, increased vascular permeability, chemotaxis of inflammatory cells, and exacerbate tissue destruction and edema [16]. These events cause the clinical findings of urticaria and or angioedema. The histology of UV lesions shows perivascular neutrophilic (or, less commonly, lymphocytic) infiltrate with fibrinoid necrosis. It indicates leucocytoclastic vasculitis with evidence of postcapillary venules endothelial cells injury, and erythrocyte extravasation. Direct immuno fluorescence study of HUVS lesions demonstrates both immune complex and complement deposition in a granular pattern in or around blood vessels in the upper dermis and a striking deposition of immunoglobulins and complement along the dermal-epidermal junction [17,18].
Evidence suggest that anti-C1q antibody in SLE patients do not bind to reduced C1q, but about 60 percent of HUVS anti-C1q Ab preparations do bind [2]. The tertiary structure of the intact C1q collagen-like domain is required for the recognition of the anti-C1q antibodies in SLE, whereas HUVS precipitins bind epitopes on reduced and denatured C1q [19].
When disease is active, all the patients with HUVS are hypocomplementemic with low C1q levels and detectable anti-C1q ab levels. C3 and C4 levels fluctuate from undetectable to normal low. The remission of HUVS, whether spontaneous or drug induced, is associated with normalization of C3 and C4, but C1q may remain slightly decreased even when disease is inactive [2,20]. C1q has been shown to be involved in both the activation and inhibition of T cells. Anti-C1q may interfere with the clearance of apoptotic cells, influencing induction and expression of autoimmunity [21].
C1q precipitins (C1q-p also referred to as anti-C1qAb/C1q) are IgG autoantibodies that bind to the Fc portion, collagen-like regions, of the C1 molecule. They form immune complexes and activate the complement system (C3a and C5a) by the classical pathway within and around blood vessels [15]. The activation of this cascade causes mast cell degranulation and generates anaphylatoxins. In addition, it upregulates chemokines and cytokines, which lead to deposition of immune complexes, increased vascular permeability, chemotaxis of inflammatory cells, and exacerbate tissue destruction and edema [16]. These events cause the clinical findings of urticaria and or angioedema. The histology of UV lesions shows perivascular neutrophilic (or, less commonly, lymphocytic) infiltrate with fibrinoid necrosis. It indicates leucocytoclastic vasculitis with evidence of postcapillary venules endothelial cells injury, and erythrocyte extravasation. Direct immuno fluorescence study of HUVS lesions demonstrates both immune complex and complement deposition in a granular pattern in or around blood vessels in the upper dermis and a striking deposition of immunoglobulins and complement along the dermal-epidermal junction [17,18].
Evidence suggest that anti-C1q antibody in SLE patients do not bind to reduced C1q, but about 60 percent of HUVS anti-C1q Ab preparations do bind [2]. The tertiary structure of the intact C1q collagen-like domain is required for the recognition of the anti-C1q antibodies in SLE, whereas HUVS precipitins bind epitopes on reduced and denatured C1q [19].
When disease is active, all the patients with HUVS are hypocomplementemic with low C1q levels and detectable anti-C1q ab levels. C3 and C4 levels fluctuate from undetectable to normal low. The remission of HUVS, whether spontaneous or drug induced, is associated with normalization of C3 and C4, but C1q may remain slightly decreased even when disease is inactive [2,20]. C1q has been shown to be involved in both the activation and inhibition of T cells. Anti-C1q may interfere with the clearance of apoptotic cells, influencing induction and expression of autoimmunity [21].
CLINICAL FEATURES
In HUV, the main clinical feature is recurrent to chronic UV. Half of the patients experience angioedema, involving the lips, tongue, periorbital regions and hands. In some cases, angioedema can be the first sign of HUS and HUVS. The wheals last more than 24 hours and hyperpigmentation of the skin appears when the wheals resolve. The lesions may be painful, cause a burning sensation, and some may have itching. In addition to skin findings, the patients with HUVS might have extracutaneous manifestations including constitutional symptoms (fever, malaise, and fatigue); musculoskeletal symptoms; ocular inflammation such as conjunctivitis, episcleritis, and uveitis; serositis; obstructive lung disease; Raynaud’s phenomenon; renal disease; gastrointestinal symptoms; cardiac involvement; and various neurologic problems.
Arthralgia and arthritis affecting various joints are also common findings. These symptoms are mainly seen in up to 50% of patients with systemic manifestations of HUVS. Typically, the elbows, wrists, knees, and ankles are involved and the joint pain is often transient. Joint deformities can be observed [22]. Moderate to severe chronic obstructive pulmonary disease and asthma, seen in 20% to 50% of patients [2,23]. Renal disease occurs in up to 50% of patients with HUVS and is manifested by proteinuria and microscopic hematuria. Renal biopsy findings may show proliferative Glomerulonephritis (GN), crescentic GN, membrano proliferative glomerulonephritis, focal necrotizing vasculitis, or tubulointerstitial nephritis. The pattern of renal findings is indistinguishable from SLE nephropathy. The renal involvement seems to be more severe in children and rapid progression to end stage renal disease has occurred. In these cases, the renal disease responded well to the immunosuppressive treatment [11,24,25].
Arthralgia and arthritis affecting various joints are also common findings. These symptoms are mainly seen in up to 50% of patients with systemic manifestations of HUVS. Typically, the elbows, wrists, knees, and ankles are involved and the joint pain is often transient. Joint deformities can be observed [22]. Moderate to severe chronic obstructive pulmonary disease and asthma, seen in 20% to 50% of patients [2,23]. Renal disease occurs in up to 50% of patients with HUVS and is manifested by proteinuria and microscopic hematuria. Renal biopsy findings may show proliferative Glomerulonephritis (GN), crescentic GN, membrano proliferative glomerulonephritis, focal necrotizing vasculitis, or tubulointerstitial nephritis. The pattern of renal findings is indistinguishable from SLE nephropathy. The renal involvement seems to be more severe in children and rapid progression to end stage renal disease has occurred. In these cases, the renal disease responded well to the immunosuppressive treatment [11,24,25].
TREATMENT
Patients with normal complement levels have a self-limited disease and require only antihistamines when cutaneous lesions develop. Treatment of HUV requires therapy with steroids and immuno suppressives as patients only get temporary relief with the use of antihistamines. There is no specific treatment for HUVS and the consensus for effective therapy has not been established yet. Patients presenting with only cutaneous disease and arthralgia without any major organ involvement may be managed with low-dose prednisone, hydroxychloroquine, or dapsone. On the other hand, patients with major organ involvement, such as glomerulonephritis, may require high doses of corticosteroids and cytotoxic agents similar to the treatment for active SLE. Response to treatment can be monitored by a decrease in circulating anti-C1q titer and normalization of C3 and C4 levels. However, despite treatment, C1q often remains low. Cyclosporin A, has shown good results in HUVS, particularly for renal and pulmonary disease. Mycophenolate mofetil has been beneficial for maintenance therapy in a patient of HUVS after treatment with glucocorticoids and cyclophosphamide [26]. Another patient with HUVS and SLE that was refractory to mycophenolate mofetil, high-dose methylprednisolone, and intravenous immunoglobulin, was successfully treated with rituximab [27]. Plasma exchange and intravenous γ-globulins may also be of value in patients with renal involvement [25,28]
The prognosis for HUV depends on the severity of systemic involvement like pulmonary, cardiac and/or renal disease. When present, pulmonary disease is the major cause of death. Acute laryngeal edema can be life-threatening. Although uncommon in childhood, the prognosis is worse for early onset HUV patients because of more frequent and severe renal involvement.
The prognosis for HUV depends on the severity of systemic involvement like pulmonary, cardiac and/or renal disease. When present, pulmonary disease is the major cause of death. Acute laryngeal edema can be life-threatening. Although uncommon in childhood, the prognosis is worse for early onset HUV patients because of more frequent and severe renal involvement.
DISCUSSION
Hypocomplementemic urticarial vasculitis is an entity that describes patients with UV and hypocomplementemia who do not meet diagnostic criteria for HUVS. Most of the time, patients with HUV have cutaneous disease and few or no systemic manifestations.
It is a rare autoimmune disorder consistent with recurrent urticarial vasculitis that causes a stinging or burning sensation on the skin, low complement levels usually during rash outbreaks, arthralgia/arthritis, and eye inflammation [29]. The complete pathogenesis of this syndrome is unknown. Possible pathologies include immune complex mediated via C1q- precipitin, T-lymphocyte response, and anti-C1q- antibody mediated [22].
As in our patient’s presentation, arthralgia and arthritis of several joints occurs in about 50% of cases. The arthralgia and arthritis is migratory and typically occurs in larger joints such as the elbows, wrists, knees, and ankles. In the systemic form, obstructive lung disease, angioedema, ocular inflammation, and glomerulonephritis are common manifestations. The spectrum of renal involvement is wide ranging from no to mild involvement to patients requiring dialysis. Screening urinalysis should be done when the diagnosis is suspected or when the patient has clinical symptoms. Unfortunately, renal involvement can be more severe in children [22,30]. In fact, the biopsy findings in these patients are indistinguishable from SLE [31]. Lung involvement may be non-existent at the time of presentation. However, these patients can develop dyspnea, cough, hemoptysis, pleural effusions, and even COPD. C1q precipitins may be associated with increased tendency for obstructive lung disease [22].
Normocomplementemic patients usually have minimal or no systemic involvement, and often have a better prognosis. However, hypocomplementemic patients might have more severe multi-organ involvement. The patients presenting with urticarial vasculitis that is non-necrotizing are treated with antihistamines, dapsone, colchicine, hydroxychloroquine, or indomethacin, but corticosteroids are often required. With necrotizing skin lesions or visceral involvement, corticosteroids are usually indicated. The patients with severe urticarial vasculitis who are resistant to corticosteroid treatment, may require other immunosuppressive agents such as azathioprine, cyclosporine, and cyclophosphamide or mycophenolate mofetil.
It is a rare autoimmune disorder consistent with recurrent urticarial vasculitis that causes a stinging or burning sensation on the skin, low complement levels usually during rash outbreaks, arthralgia/arthritis, and eye inflammation [29]. The complete pathogenesis of this syndrome is unknown. Possible pathologies include immune complex mediated via C1q- precipitin, T-lymphocyte response, and anti-C1q- antibody mediated [22].
As in our patient’s presentation, arthralgia and arthritis of several joints occurs in about 50% of cases. The arthralgia and arthritis is migratory and typically occurs in larger joints such as the elbows, wrists, knees, and ankles. In the systemic form, obstructive lung disease, angioedema, ocular inflammation, and glomerulonephritis are common manifestations. The spectrum of renal involvement is wide ranging from no to mild involvement to patients requiring dialysis. Screening urinalysis should be done when the diagnosis is suspected or when the patient has clinical symptoms. Unfortunately, renal involvement can be more severe in children [22,30]. In fact, the biopsy findings in these patients are indistinguishable from SLE [31]. Lung involvement may be non-existent at the time of presentation. However, these patients can develop dyspnea, cough, hemoptysis, pleural effusions, and even COPD. C1q precipitins may be associated with increased tendency for obstructive lung disease [22].
Normocomplementemic patients usually have minimal or no systemic involvement, and often have a better prognosis. However, hypocomplementemic patients might have more severe multi-organ involvement. The patients presenting with urticarial vasculitis that is non-necrotizing are treated with antihistamines, dapsone, colchicine, hydroxychloroquine, or indomethacin, but corticosteroids are often required. With necrotizing skin lesions or visceral involvement, corticosteroids are usually indicated. The patients with severe urticarial vasculitis who are resistant to corticosteroid treatment, may require other immunosuppressive agents such as azathioprine, cyclosporine, and cyclophosphamide or mycophenolate mofetil.
CONCLUSION
We reported a case of a 6 year old female with HUV that responded well to oral prednisolone. HUV is a rare autoimmune disorder seen in children and consistent with consistent recurrent urticarial vasculitis that causes non-itchy painful, hyperpigmented macules and wheals, low complement levels during rash outbreaks, arthralgia/arthritis, and eye inflammation [31]. We reviewed the current understanding of the pathophysiology of this disease, briefly discussed the clinical manifestations, differential diagnosis and summarized the current treatment regimens.
REFERENCES
- Davis MD, Brewer JD (2004) Urticarial vasculitis and hypocomplementemic urticarial vasculitis syndrome. Immunol Allergy Clin North Am 24: 183-213.
- Wisnieski JJ (2000) Urticarial vasculitis. Curr Opin Rheumatol 12: 24-31.
- Black AK (1999) Urticarial vasculitis. Clin Dermatol 17: 565-569.
- Martini A, Ravelli A, Albani S, De Benedetti F, Massa M, et al. (1994) Hypocomplementemic utricularial vasculitis syndrome with severe systemic manifestations. J Pediatr 124: 742-744.
- Waldo FB, Leist PA, Strife CF, Forristal J, West CD (1985) Atypical hypocomplementemic vasculitis syndrome in a child. J Pediatr 106: 745-750.
- Cadnapaphornchai MA, Saulsbury FT, Norworrd VF (2000) Hypocomplementemic urticarial vasculitis: report of a pediatric case. Pediatr Nephrol 14: 328-331.
- Renard M, Wouters C, Proesmans W (1998) Rapidly progressive glomerulonephritis in a boy with hypocomplementaemic urticarial vasculitis. Eur J Pediatr 157: 243-245.
- Wisnieski J, Baer AN, Christensen J, Cupps TR, Flagg DN, et al. (1995) Hypocomplementemic urticarial vasculitis syndrome. Clinical and serological findings in 18 patients. Medicine (Baltimore) 74: 24-41.
- Filosto M, Cavallaro T, Pasolini G, Broglio L, Tentorio M, et al. (2009) Idiopathic hypocomplementemic urticarial vasculitis-linked neuropathy. J Neurol Sci 284: 179-181.
- Firestein GS, Budd RC, Harris ED, McInnes IB, Ruddy S, et al. (2008) Kelly’s Textbook of Rheumatology. 8th ed. Saunders, Philadelphia, PA, USA.
- Balsam L, Karim M, Miller F, Rubinstein S (2008) Crescentic glomerulonephritis associated with hypocomplementemic urticarial vasculitis syndrome. Am J Kidney Dis 52: 1168-1173.
- DeAmicis T, Mofid MZ, Cohen B, Nousari HC (2002) Hypocomplementemic urticarial vasculitis: report of a 12-year-old girl with systemic lupus erythematosus. J Am Acad Dermatol 47: 273-274.
- O’Donnell B, Black A (1995) Urticarial vasculitis. Int Angiol 14: 166-174.
- Mehregan DR, Hall MJ, Gibson LE (1992) Urticarial vasculitis: a histopathologic and clinical review of 72 cases. J Am Acad Dermatol 26: 441-448.
- Chew G, Gatenby P (2007) Inflammatory myositis complicating hypocomplementemic urticarial vasculitis despite on-going immunosuppression. Clin Reumatol 26: 1370-1372.
- Jara L, Navarro C, Medina G, Vera-Lastra O, Saavendra MA (2009) Hypocomplementemic urticarial vasculitis syndrome. Curr Rheumatol Reports 11: 410-415.
- Aydogan K, Karadogan S, Adim SB, Tunali S (2006) Hypocomplementemic urticarial vasculitis: a rare presentation of systemic lupus erythematosus. Int J Dermatol 45: 1057-1061.
- Fortson J, Zone JJ, Hammond ME, Groggel GC (1986) Hypocomplementemic urticarial vasculitis syndrome responsive to dapsone. J Am Acad Dermatol 15: 1137-1142.
- Uwatoko S, Mannik M (1988) Low-molecular weight C1q-binding immunoglobulin G in patients with systemic lupus erythematosus consists of autoantibodies to the collagen-like region of C1q. J Clin Invest 82: 816-824.
- Khasnis A, Langford CA (2009) Update on vasculitis. J Allergy Clin Immunol 123: 1226-1236.
- Kallenberg CG (2008) Anti-C1q autoantibodies. Autoimmun Rev 8: 612-615.
- Buck A Jim Christensen, Morgan McCarty (2012) Hypocomplementemic Urticarial Vasculitis Syndrome. J Clin Aesthet Dermatol 5: 36-46.
- Venzor J, Lee WL, Huston DP (2002) Urticarial vasculitis. Clin Rev Allergy Immunol 23: 201-216.
- Ramirez G, Saba SR, Espinoza L (1987) Hypocomplementemic vasculitis and renal involvement. Nephron 45: 147-150.
- Grimbert P, Schulte K, Buisson C, Desvaux D, Baron C, et al. (2001) Renal transplantation in a patient with hypocomplementemic urticarial vasculitis syndrome. Am J Kidney Dis 37: 144-148.
- Worm M, Sterry W, Kolde G (2000) Mycophenolate mofetil is effective for maintenance therapy of hypocomplementaemic urticarial vasculitis. Br J Dermatol 143: 1324-1326.
- Saigal K, Valencia IC, Cohen J, Kerdel FA (2003) Hypocomplementemic urticarial vasculitis with angioedema, a rare presentation of systemic lupus erythematosus: rapid response to rituximab. J Am Acad Dermatol 49: 283-285.
- Staubach-Renz P, von Stebut E, Bräuninger W, Maurer M, Steinbrink K (2007) [Hypocomplementemic urticarial vasculitis syndrome. Successful therapy with intravenous immunoglobulins]. Hautarzt 58: 693-697.
- Al Mosawi ZSA, Al Hermi BEA (2013) Hypocomplementemic Urticarial Vasculitis Syndrome in an 8-year-old Boy: A Case Report and Review of Literature. Oman Med J 28: 275-277.
- Pasini A, Bracaglia D, Aceti A, Vivarelli M, Lavacchini A, et al. (2014) Renal involvement in hypocomplementaemic urticarial vasculitis syndrome: a report of three paediatric cases. Rheumatology (Oxford) 53: 1409-1413.
- Jara LJ, Navarro C, Medina G, Vera-Lastra O, Saavedra MA (2009) Hypocomplementemic urticarial vasculitis syndrome. Curr Rheumatol Rep 11: 410-415.
Citation: Dokmeci E, Camacho JE, Dokmeci OC (2015) Hypocomplementemic Urticarial Vasculitis in a Pediatric Patient. J Neonatol Clin Pediatr 2: 008.
Copyright: © 2015 Elif Dokmeci, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
© 2026, Copyrights Herald Scholarly Open Access. All Rights Reserved!