Incontinentia Pigmenti in a 3-Year-Old Female: A Case Report
*Corresponding Author(s):
Anjali MishraSchool Of Medicine, Creighton University, Omaha, NE 68178, United States
Tel:+1-925-895-8905,
Email:anjalimishra@creighton.edu
Abstract
Incontinentia Pigmenti (IP), also known as Bloch-Sulzberger syndrome, is a rare X-linked genetic disorder characterized by a distinctive cutaneous manifestation along with variable systemic involvement. We present a case of a 3-month-old female infant diagnosed with IP, highlighting the clinical features, diagnostic approach, and management strategies. Early recognition and multidisciplinary care are crucial for optimizing outcomes in affected infants.
Keywords
Incontinentia Pigmenti; Pediatric Dermatology; Pediatrics
Background
Incontinentia Pigmenti (IP) is a rare X-linked dominant genetic disorder with an estimated incidence of 1 in 40,000 live births. It is a systemic disease caused by mutations in the IKBKG gene (also known as NEMO) located on Xq28. [1] Mutations in this gene disrupt the normal function of NEMO, diminishing its ability to modulate NF-kB. This leads to multiple downstream effects involving regulation of inflammation, cell survival, distribution of melanin, and other tissue development. IP almost exclusively affects females, with non-lethal NEMO mutations in hemizygous XY male patients being extremely rare. [2] The disorder is characterized by cutaneous, dental, ocular, and neurological abnormalities due to mutations primarily affecting tissues derived from mesoderm and ectoderm during development. Skin manifestations typically present in infancy appearing in distinct stages. Dental abnormalities, such as delayed eruption or missing teeth, are also common. According to a comprehensive literature review from 1947 to 2014, 36.5% of patients with IP have eye pathology and up to 90% of those have retinal abnormalities. [3] CNS involvement may include developmental delays or intellectual disabilities. Neurologic impairments have been reported in 30% of cases. Treatment for IP is primarily focused on managing symptoms and complications that may arise, typically requiring an interdisciplinary medical approach for comprehensive care. Genetic counseling may also be necessary for affected families, given the inheritance pattern of IP. Herein, we describe a case of IP in a 3-month-old female infant.
Case Report
A 3-month-old female infant was presented to the clinic for a well-child visit. She was born full-term with no history of gestational complications. The infant’s medical history was significant for carrier status of methionine adenosyltransferase 1 alpha deficiency. Physical examination of the infant revealed cutaneous lesions with a striking distribution and pattern. Lesions were present on the limbs, trunk, and face, appearing as whorls or streaks of hyperpigmented and hypopigmented patches. Given the clinical presentation, a skin biopsy was performed on one of the hyperpigmented patches. Histopathological examination showed hyperkeratosis, acanthosis, and melanin incontinence, consistent with a diagnosis of IP. The child was found to have a mutation in the IKBKG (NEMO) gene, confirming a diagnosis of IP.
Following diagnosis, the patient was managed with a multidisciplinary approach involving ophthalmology, neurology, and dentistry. She began ophthalmologic follow-up every 6 months to closely monitor for retinal and non-retinal ocular complications associated with IP. At 19 months of age, the patient began experiencing seizure-like activities. These episodes involved unresponsive staring, loss of consciousness, stiffness and weakness of the limbs, and immediate loss of speech followed by prolonged postictal fatigue. The patient’s EEG report displayed spiky sleep architecture with possible epileptiform discharges, confirming seizure-like activity. The patient was started on Keppra 10 mg for management of her seizures and closely followed by neurology. The patient was also regularly seen by dentistry to monitor tooth development for malformation and hypodontia as she continued to grow. Overall, timely intervention and multidisciplinary support from multiple clinical specialties allowed for significant improvement of the quality of life for this patient and her family.
Discussion
IP is an X-linked disorder caused by mutations of the Xq28 locus of the IKBKG (inhibitor of kappa B kinase gamma) gene, also known as NEMO (NF-kappa-B essential modulator). [4] The IKBKG gene plays a crucial role in the NF-kappa-B signaling pathway, which is involved in the regulation of various cellular processes, including inflammation, immunity, and cell survival. Mutations disrupting the normal function of the NF-kappa B pathway lead to the characteristic features of IP. Skin manifestations observed in individuals diagnosed with IP characteristically manifest along the lines of Blaschko, which follow the migration pattern of embryonic cells during development. [5] While usually lethal in males, females can survive as a result of X-inactivation mosaicism [6].
IP follows a typical four-stage course: inflammatory or vesiculobullous (stage 1), verrucous (stage 2), hyperpigmented (stage 3), and hypopigmented (stage 4). (Stage 1) tends to occur within the first few weeks of life, and is characterized by 1-mm to 1-cm blistering lesions distributed linearly along the lines of Blaschko. Following initial presentation, the vesicles and bullae can progress to pustules and gradually regress by four months of age. As the condition progresses to (stage 2), blisters may transform into verrucous, wart-like growths or plaques on the skin of the extremities and trunk. (Figure 1) These lesions tend to appear within two to six weeks and resolve by six months of age. (Stage 3) typically occurs in childhood and is characterized by hyperpigmented streaks, whorls, or patches accompanied by atrophy. The areas of hyperpigmentation range from brown to black in coloration and can be found along lines of Blaschko. The hyperpigmentation usually regresses during adolescence, but may persist into later stages of life, particularly in the axillae and groin regions. The final stage (stage 4) is characterized by hypopigmented streaks or patches, atrophy, and absence of hair. These patches are often irregularly shaped and likewise follow Blaschko lines, mostly being reported on the lower extremities. The cutaneous findings observed in this later stage may also persist into adulthood.
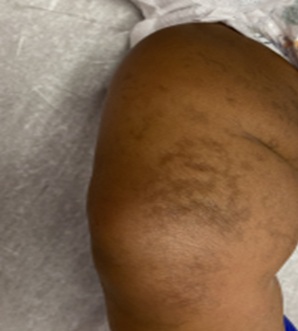 Figure 1: Patient’s anterior right thigh and knee demonstrating whorled streaks of hyperpigmentation on the skin.
Figure 1: Patient’s anterior right thigh and knee demonstrating whorled streaks of hyperpigmentation on the skin.
In addition to cutaneous manifestations, IP is characterized by a variety of ocular abnormalities, neurologic manifestations, and dental or skeletal abnormalities. [7] Patients with IP commonly experience dental anomalies such as hypodontia (missing teeth) and misshapen teeth, leading to potential oral health challenges. Eye complications in these patients are frequent, including retinal detachment and strabismus, which can adversely affect visual function and necessitate ophthalmic interventions. Furthermore, involvement of the nervous system may encompass developmental delays, intellectual disabilities, and seizures, highlighting the diverse range of complications associated with IP that require multidisciplinary medical management and care.
Conclusion
In conclusion, we present a case of Incontinentia Pigmenti, a rare and complex genetic disorder, and highlight the intricate and multisystem nature of this condition. While caught early in this patient, there is a wide spectrum of manifestations associated with this condition, ranging from distinctive skin changes to dental anomalies and central nervous system involvement. The utilization of genetic testing to identify mutations in the IKBKG gene not only aided in confirming the diagnosis but also highlighted the underlying genetic basis of the disorder. From this case, we are reminded of the importance of a multidisciplinary approach in managing individuals with IP. Collaborative efforts among dermatologists, pediatricians, geneticists, ophthalmologists, and other specialists are crucial in addressing the diverse array of symptoms and providing comprehensive care. Furthermore, this case underscores the significance of genetic counseling for affected families, offering them informed insights into the inheritance pattern, potential recurrence risks, and avenues for supportive interventions. This report contributes to the growing body of literature surrounding IP, emphasizing the necessity of ongoing research, awareness, and compassionate care for patients and families impacted by this rare genetic disorder.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Source of Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Statement of Ethics
This material has not been published in whole or in part elsewhere. Informed consent is not applicable given detailed patient identifiers are not presented.
References
- Berlin AL, Paller AS, Chan LS (2002) Incontinentia pigmenti: A review and update on the molecular basis of pathophysiology. J Am Acad Dermatol. 47:169-190.
- Song JY, Na CH, Chung BS, Choi KC, Shin BS (2008) A case of a surviving male infant with incontinentia pigmenti. Annals of dermatology. 20: 134-137.
- Swinney CC, Han DP, Karth PA (2015) Incontinentia pigmenti: A comprehensive review and update. Ophthalmic surgery, lasers & imaging. 46: 650-657.
- Poziomczyk CS, Recuero JK, Bringhenti L, Maria SFD, Campos CW, et al. (2014) Incontinentia pigmenti. An Bras Dermatol. 89: 26-36.
- Scheuerle AE, Ursini MV, Adam MP, Mirzaa GM, Pagon RA (1993) Incontinentia pigmenti. University of Washington, Seattle, Seattle (WA).
- Greene-Roethke C (2017) Incontinentia pigmenti: A summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. Journal of Pediatric Health Care. 31: e45-e52.
- Gianfaldoni S, Tchernev G, Wollina U, Lotti T (2017) Incontinentia pigmenti: A case report of a complex systemic disease. Open Access Macedonian Journal of Medical Sciences. 5: 501-505.
Citation: Mishra A, Taylor M, Kauper R, Kubesh M (2023) Incontinentia Pigmenti in a 3-Year-Old Female: A Case Report J Clin Stud Med Case Rep 10:188
Copyright: © 2023 Anjali Mishra, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.