Karyomegalic Interstitial Nephritis-A Rare Cause of Chronic Tubulointerstitial Nephritis
*Corresponding Author(s):
Kanishk GuptaDepartment Of Pathology, GIPMER, New Delhi, India
Tel:+91 7300582485,
Email:kanishk1gupta@gmail.com
Abstract
Karyomegalic Interstitial Nephritis (KIN) is a rare disease, which usually presents with slowly progressive chronic kidney disease, eventually leading to end stage renal disease in early adulthood. Histological findings consist of enlarged and hyperchromatic nuclei in scattered tubular epithelial cells throughout the nephron accompanied by interstitial fibrosis around atrophic tubules. Herein, we report a case of a 24-year-old femalewho presented with stage 3 chronic kidney disease. Renal biopsy revealed chronic tubulointerstitial nephritis and an unusually marked karyomegaly particularly of the tubular epithelium.
Keywords
Chronic tubulointerstitial nephritis; Karyomegaly; KIN
INTRODUCTION
KIN is a rare diseasecharacterized by chronic tubulointerstitial nephritis associated with large tubular epithelial cell nuclei.It was first described by Burry et al. [1] and subsequently named by Mihatsch et al. [2]. This disease has no known treatment and progresses to chronic kidney disease in a short span of time [2]. Extra renal manifestations are uncommon and consists of recurrent respiratory tract infections and transient elevations in liver function tests. The diagnosis has been linked to mutations in the FAN1 (FANCD2/FANCI-Associated Nuclease 1) gene, a gene involved in DNA damage response pathway [3]. Here we report a case of a 24 year old female who presented with stage 3 CKD due to KIN.
CASE REPORT
A 24-year-old female was referred to our nephrology department of our hospital with impaired renal function. She presented with stage 3 chronic kidney disease and had history of hematuria for 6 years. Serum creatinine was 2.5 mg/dl and 24 hr urinary protein was 1.4 g at the time of biopsy. There was no history of significant drug ingestion including exposure to mycotoxins, viral infections, environmental or agricultural toxins. Her liver enzymes were also normal.
Renal biopsy (Figure 1-5) was done to evaluate the case. There were two cores of renal tissue with 10 glomeruli. Seven glomeruli were sclerosed. The viable glomeruli were unremarkable. The tubules had a denuded lining with marked nuclear changes. There was nucleomegaly (karyomegaly), alteration of chromatin texture, hyperchromasia, nuclear pleomorphism and occasional multinucleation. There was mild tubular atrophy involving 15 to 20 % of the cortex. The interstitium had a mild lymphomononuclear infiltrate. The blood vessels showed hyalinosis. Immunoflourescence showed no immune deposits for IgG, IgM, IgA, C3, C1q, kappa and lambda. Immunohistochemistry for SV 40 and CMV were negative. The Ki-67 stain showed virtually no positive cells. A diagnosis of KIN was made.
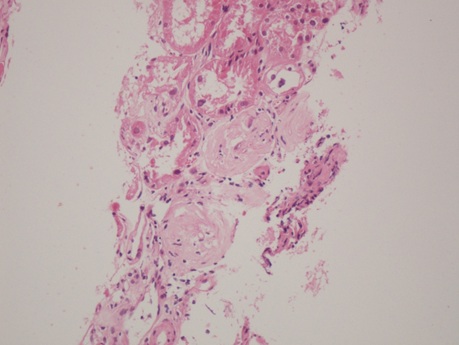
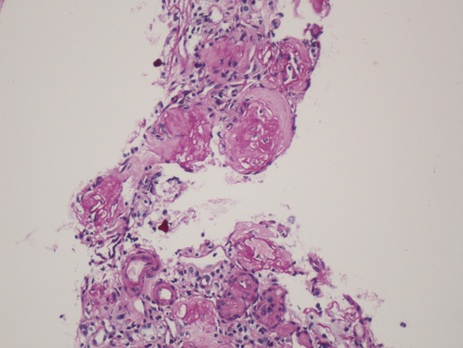
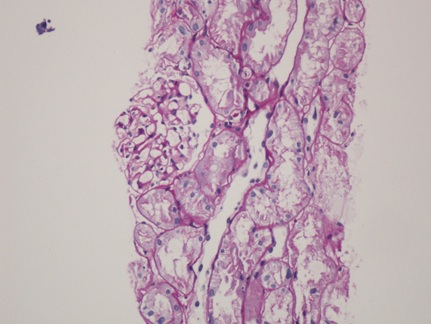 Figure (1-3): Low power view showing glomerular obsolescence, tubular atrophy and tubular epithelial cell nuclei changes.(100x).
Figure (1-3): Low power view showing glomerular obsolescence, tubular atrophy and tubular epithelial cell nuclei changes.(100x).
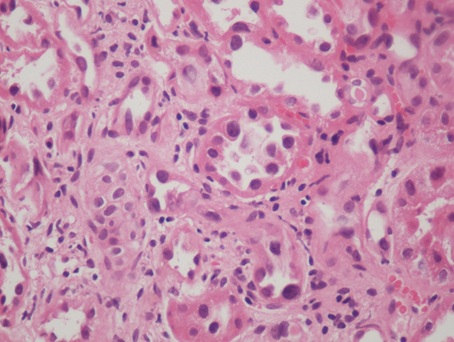
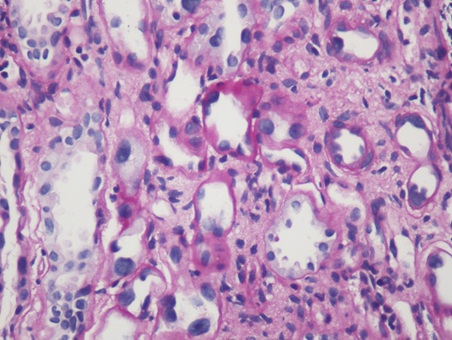 Figure 4, 5: High power view showing karyomegaly, hyperchromasia and nuclear pleomorphism of tubular epithelial cell nuclei changes.(400x).
Figure 4, 5: High power view showing karyomegaly, hyperchromasia and nuclear pleomorphism of tubular epithelial cell nuclei changes.(400x).
DISCUSSION
KIN is a rare cause of hereditary chronic interstitial nephritis described first time over 40 years ago.In 1974, Burry et al. [1], reported the case of a young woman dying from hepatocellular carcinoma and who displayed dysplastic nuclei in the renal epithelium.KIN term was introduced by Mihatsch et al. [2], who described 3 cases of systemic karyomegaly associated with chronic interstitial nephritis. The prevalence of KIN is less than 1% of all biopsies examined, with <50 cases reported till date in the literature. It presents at 9 to 51 years of age (median: 33 years) [4].
KIN usually presents with a slowly progressive chronic kidney disease, eventually leading to end stage renal disease at 30 to 40 years of age [4]. Patients show asymptomatic proteinuria, usually <1 g/day. More than 75% patients also show glycosuria and less than one third can present along with hematuria [5]. Extrarenal manifestations are rare. They may include recurrent respiratory tract infections and elevated liver enzymes, which were not present in our case. Case study by N. Bennani Guesbessi [6], showed a long history of recurrent respiratory tract infection episodes while the study by Ebru Uz et al. [7], showed slight elevation of liver enzymes. 50% have a family history of renal disease [4]. Karyomegalic cells have been identified in various tissues including astrocytes, schwann cells, intestinal smooth muscle, acinar cells of pancreas, Kupffer cells of liver and bile duct epithelium, though not diagnostic.
Renal biopsy shows karyomegaly in scattered tubules throughout the nephron especially in proximal tubules. Nuclei are 2 to 5 times larger than normal with hyperchromasia, and no mitotic figures. It is usually associated with global or segmental glomerulosclerosis and tubular atrophy accompanied by interstitial fibrosis in affected areas. Rarely, karyomegaly can be seen in glomerular cells and smooth muscles of blood vessels. Electron microscopy shows abnormal nuclei with expanded loose matrix and convoluted nuclear membranes. Immunohistochemistry for Ki67 and PCNA do not stain enlarged nuclei, as they are not in cell cycle. The high DNA ploidy values are indicative of increased degree of karyotypic abnormalities and are recognized as markers of malignant potential and/or poor prognosis in a number of diseases [8]. Urine cytology shows karyomegalic cells in urine which can be misinterpreted as carcinoma.4KIN can have other differential diagnoses, including viral infections detected by IHC for viral antigens, chemotherapy such as alkylating agents (ifosfamide), radiotherapy, exposure to heavy metals and mycotoxins, particularly Ochrotoxin A. The above conditions have less karyomegaly, a more restricted distribution across the nephron and a higher Ki 67 index [9].
Historically, KIN was thought to be a hereditary disorder because almost half of patients had a family history of nephropathy. It has been recently ascribed to autosomal recessive mutations in the FAN1 gene, which encodes for Fanconi anemia associated nuclease 1. Nuclease belongs to the Fanconi anemia DNA damage response pathway and is required for repair of DNA interest and cross links [10]. FAN1 mutations are predominantly expressed in kidney, liver and neuronal cells [9]. FAN1 mutations increases susceptibility to DNA toxins [4].
In summary, KIN is a rarely seen important condition and probably underdiagnosed disorder characterized by large tubular epithelial cell nuclei. It is important to diagnose this condition, as it is a progressive disorder that causes irreversible chronic damage.KIN should always be kept as a differential diagnosis in a young patient with symptoms of renal dysfunction, active urine sediment and abnormal liver enzymes in the absence of other environmental factors associated with interstitial nephritis.
REFERENCES
- Burry AF (1974) Extreme dysplasla in renal epithelium of a young woman dying from hepatocarcinoma. J Pathol 113: 147-150.
- Mihatsch MJ, Gudat F, Zollinger HU, Heierli C, Thölen H, et al. (1979) Systemic karyomegaly associated with chronic interstitial nephritis. A new disease entity? Clin Nephrol 12: 54-62.
- Isnard P, Rabant M, Labaye J, Antignac C, Knebelmann B, Zaidan M (2016) Karyomegalic interstitial nephritis: A case report and review of the literature. Medicine (Baltimore) 95: 3349.
- Colvin RB, Chang AC (2019) Diagnostic Pathology: Kidney Diseases. Elsevier, Philadelphia, USA.
- Monga G, Banfi G, Salvadore M, Amatruda O, Bozzola C, et al. (2006) Karyomegalic interstitial nephritis: Report of 3 new cases and review of the literature. Clin Nephrol 65: 349-355.
- Bennani Guebessi N, Karkouri M (2016) Karyomegalic interstitial nephritis. CEN Case Rep 5: 23-25.
- Uz E, Bayram Y, Haltas H, Bavbek N, Kanbay M, et al. (2001) Karyomegalic tubulointerstitial nephritis: A rare cause of chronic kidney disease. Nephrourol 3: 201-203.
- Vinay KS, Siddappa S (2017) Karyomegalic interstitial nephritis: A review of literature. BAOJ Urol Nephrol 1: 005.
- Lachaud C, Slean M, Marchesi F, Lock C, Odell E, et al. (2016) Karyomegalic interstitial nephritis and DNA damage-induced polyploidy in Fan1nuclease-defective knock in mice. Genes Dev 30: 639-644.
- Zhou W, Otto EA, Cluckey A, Airik R, Hurd TW, et al. (2012) FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat Genet 44: 910-915.
Citation: Gupta K, Swain M, Gowrishankar S (2020) Karyomegalic Interstitial Nephritis-A Rare Cause of Chronic Tubulointerstitial Nephritis. J Nephrol Renal Ther 6: 042.
Copyright: © 2020 Kanishk Gupta, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.