Microrna Associated with Generation, Development, and Maintenance of Atrial Fibrillation
*Corresponding Author(s):
Hui GongDepartment Of Internal Medicine, Division Of Cardiology, Jinshan Hospital, Shanghai, China
Tel:+862134189990,
Fax:+862157039502
Email:guoxiong.xu@fudan.edu.cn
Lubna Nadeem
Lunenfeld Tanenbaum Research Institute, Mount Sinai Hospital, Toronto, Canada
Tel:+14168900073,
Email:nadeem@lunenfeld.ca
Guoxiong Xu
Center Laboratory, Jinshan Hospital, Fudan University, Shanghai, China
Tel:+8613918566212,
Fax:+862167226910
Email:liyuanhn@aliyun.com
Abstract
MicroRNAs (miRNAs) are a class of small non-coding RNAs, playing important roles in the regulation of the cardiovascular function. MiRNAs are associated with the generation, development, and maintenance of atrial fibrillation through the regulation of the gene expression on targets of ion channels, transporters, calcium binding proteins, extracellular matrix proteins, and other factors. Understanding the relationship between miRNAs and atrial fibrillation is critical to identifying the diagnostic markers, prognosis and therapeutic targets.
Keywords
ABBREVIATIONS
AF - Atrial Fibrillation
Ang II - Angiotensin II
APD - Action Potential Duration
AT1-R - Angiotensin II Type 1 Receptor
CACNA1C - Calcium Channel Voltage-Dependent L Type Alpha 1C Subunit
CACNB1 - Calcium Channel Voltage-Dependent Beta 1 Subunit
CaMKII - Calcium/Calmodulin-Dependent Protein Kinase II
COL1A1 - Collagen Type I Alpha 1
COL3A1 - Collagen Type III Alpha 1
CTGF - Connective Tissue Growth Factor
DGCR8 - DiGeorge Syndrome Critical Region 8
ERK1/2 - Extracellular Signal-Regulated Kinase1/2
ECM - Extracellular Matrix
ERP - Effective Refractory Period
GJA1 - Gap Junction Alpha 1 Protein
ICa2+ - Intracellular Calcium Current
IK1 - Inward Rectifier Potassium Current
KCNJ2 - Inwardly-Rectifying Channel, Subfamily J, Member 2
KCNN3 - Potassium intermediate/small conductance calcium-activated channel subfamily N member 3
LOX - Lysyl Oxidase
MAPK - Mitogen-Activated Protein Kinase
miR or miRNA - MicroRNA
mRNA - Messenger RNA
NF-?B - Nuclear Factor-?B
PDCD4 - Programmed Cell Death 4
PP2A - Protein Phosphatase
PTEN - Phosphatase and Tensin Homology
RAAS - Renin-Angiotensin-Aldosterone System
Rac1 - Ras-Related C3 Botulinum Toxin Substrate 1
RISC - miRNA-Induced Silencing Complex
RNA - Ribonucleic Acid
RNase - Ribonuclease
RyR2 - Ryanodine Receptor Type 2
SK3 - Small-Conductance Calcium-Activated Potassium Channel 3
Spry1 - Sprouty Homolog 1
TGF-ß - Transforming growth factor-ß
TGF-ßRII - TGF-ß Receptor Type II
THRAP1 - Thyroid Hormone-Associated Protein 1
UTR - Untranslated Region
INTRODUCTION
Atrial Fibrillation (AF) is the most common sustained arrhythmia which affects quality of life drastically [1]. The incidence of AF is 16.8% and 7.6% in men and women, respectively [2]. AF can cause and exacerbate heart failure. Also it is a significant risk factor for ischemic stroke [3,4] and therefore a major cause of cardiovascular morbidity and mortality [5]. Although a copious body of both basic and clinical research exists on AF, not all of the specific pathophysiological mechanisms of the generation, development and maintenance of AF are fully understood. Evidence shows that multiple factors are involved in the generation and development of AF. These factors include oxidative stress, systemic inflammation, autonomic imbalance, endocrine disorders, early electrical remodeling, such as shortened atrial Effective Refractory Period (ERP) and Action Potential Duration (APD) [6], and late mechanical remodeling, such as cardiac hypertrophy and myocardial fibrosis [7]. Recently a few studies have unveiled that miRNAs expression and/or deregulation can influence the generation, development and maintenance of AF. This review focuses on the current understanding about the role and function of miRNAs in AF.
BIOGENESIS AND FUNCTION OF MIRNAS
MiRNAs are highly conserved, non-coding endogenous and single-stranded RNAs, which bind to the 3'-Untranslated Regions (UTRs) of target mRNAs to regulate the expression of target genes [8]. MiRNA-mediated post transcriptional gene regulation involves the degradation of the target mRNA or the suppression of its translation depending upon the sequence complementarity between the miRNA seed-sequence and the 3'-UTR of the target mRNA. Partial binding between seed sequence and target mRNA results in the repression of translation, whereas extensive pairing complementarity leads to the degradation of target mRNA [9-11].
The multiple-to-multiple relationship between miRNAs and their targets is recognised [12]. In such relationships one miRNA can target several genes and similarly several miRNAs or a miRNA cluster can target one single gene. Generally, the biological processes of miRNA biogenesis include four steps. First, generation/transcription of primary transcript called primary-miRNA (pri-miRNA) is mediated by RNA Polymerase II in the nucleus [9], which is then polyadenylated and capped by 7-methylguanosine [13]. Second, the pri-miRNA is cleaved by Drosha, a Ribonuclease (RNase) III enzyme, with its co-activator partner, DGCR8 (DiGeorge Syndrome Critical Region 8, a microprocessor complex unit) to form precursor miRNA (pre-miRNA) [14,15]. The third step involves the cytosol export of pre-miRNA by the protein Exportin 5/Ran-GTP complex [16] and further cleavage into a mature miRNA by Dicer, another RNase III enzyme [14]. In the fourth and the last step, the double-stranded mature miRNA is dissociated and incorporated into the miRNA-Induced Silencing Complex (RISC) which then targets complementary mRNAs [15,17] (Figure 1). Typically, miRNA expression is time and tissue type-specific [18].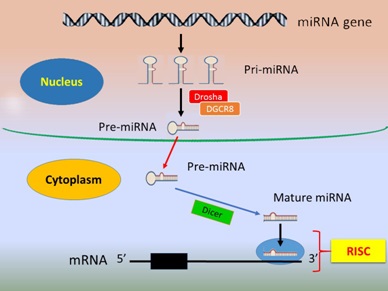
Figure 1: Biogenesis and function of miRNA.
The nucleus primary-miRNAs (pri-miRNA) are transcribed from long miRNA genes mediated by RNA polymerase II and cleaved by the Ribonuclease III (RNase III) endonuclease Drosha with its partner DGCR8 to turn into precursor-miRNAs (pre-miRNA), which then move from the nucleus to the cytoplasm where the cytoplasmic enzyme Dicer further cleaves the pre-miRNA into double-stranded mature miRNA. The mature miRNAs are incorporated into the RNA-Induced Silencing Complex (RISC). A mature miRNA binds to its target mRNA at 3'-UTR, leading to mRNA degradation or transcriptional repression.
MiRNAs are associated with many physiological processes: embryonic development, cell proliferation, differentiation, apoptosis, autophagy, and tissue or organ formation and remodeling. Especially in the heart, miRNAs play a vital role in angiogenesis, fibrosis, myocardial cells regeneration, and cardiac remodeling and therefore considered to be strong potential tools for the diagnosis and treatment of heart related diseases [9,19-21]. MiRNAs can affect cardiomyocyte excitability and conductivity by the regulation of ion channels permeability, transporters and associated regulatory proteins which can lead to different forms of arrhythmias [22]. The expression of miRNA spectrum in atrial tissue is significantly different in mitral stenosis patients with AF compared with healthy subjects [23]. AF alters the miRNA expression profiles of the left atria of mitral stenosis patients [24], suggesting that miRNA may be involved in the generation and development of AF. In fact, AF is a complex pathology, involving both primary electrical and structure factors. Different miRNA families are associated with different aspects of AF (Table1).
| miRNA | Target | Function | Possible role in AF | Reference |
| miR-1 | KCNJ2 GJA1 RyR2 | Abnormal Ca2+ handling | Atrial electrical remodeling | [25] |
| miR-19 | CTGF | Extracellular matrix, myocardial fibrosis | Atrial structural remodeling | [26] |
| miR-21 |
PDCD4 PTEN |
MMP-2 expression increase, extracellular matrix, myocardial fibrosis | Atrial structural remodeling | [27,28] |
| miR-26 | KCNJ2 | IK1 increase | Atrial electrical remodeling | [29] |
| miR-29 | COL1A1 COL3A1 | Collagen secretion, inflammation | Atrial structural remodeling | [30] |
| miR-30 | Ang II | Myocardial hypertrophy | Atrial structural remodeling | [31] |
| miR-133 | ERK1/2 TGF- ß1 TGF-ßRII | Myoblast proliferation, myogenesis | Atrial structural remodeling | [32,33] |
| miR-155 | AT1-R | Endothelial dysfunction, vascular remodeling, inflammation | Atrial structural remodeling | [34] |
| miR-208 | THRAP1 myostatin | Myocardial hypertrophy | Atrial structural remodeling | [35] |
| miR-328 | CACNA1C CACNB1 | Ica2+ reduction | Atrial electrical remodeling | [36] |
| miR-499 | KCNN3 | Abnormal Ca2+ handling | Atrial electrical remodeling | [37] |
| miR-590 | TGF- ß1 TGF-ßRII | Collagen content increase | Atrial structural remodeling | [33] |
ROLE OF MIRNAS IN THE GENERATION AND REGULATION OF AF
The mechanisms of AF are complex, but recent studies have shown that miRNAs contribute to the generation and regulation of AF in several ways (Figure 2). For example, miRNAs can induce cardiac immune/inflammatory response [38,39] and affect ion channels as well as structural and electrical remodeling including intracellular calcium overload [40]. These, in turn, can result in shortening of the action potential ERP and APD, which is conducive to the formation of reentry [4].
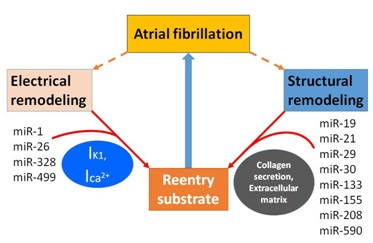
Figure 2: MiRNAs associated with atrial fibrillation.
Two mechanisms generate and develop atrial fibrillation by making substrate more prone to reentry. MiR-1, miR-26, miR-328 and miR-499 regulate atrial electrical remodeling through the modulation of ion channel (IK1, Ica2+) activity and expression. MiR-19, miR-21, miR-29, miR-30, miR-133, miR-155, miR-208 and miR-590 result in the increase of collagen secretion and extracellular matrix deposition which leads to atrial structural remolding. IK1, inward rectifier potassium current. Ica2+, intracellular calcium current.
miR-29: Role in cardiac immune response
MiR-29 has a crucial role in the generation and development of AF. The down-regulation of miR-29 can induce the fibrosis of the liver [41] and lung [42] and can also cause aneurysms [43]. The risk of AF prevalence is increased by 30% with celiac disease [44]. Overall, inflammation and chronic inflammatory diseases, such as psoriasis, also increase the risk of AF [45]. Therefore, AF is partially moderated by the humoral and systemic inflammatory response induced by local fibrosis. The miR-29 family (miR-29a, miR-29b and miR-29c) is expressed in T and B cells, dendritic cells and thymic epithelial cells, which play a regulatory role in the adaptive immune pathway [46]. Multiple biopsy specimens in left atria with AF have shown lymphocytic myocarditis in 66% of patients and noninflammatory localized cardiomyopathy in 17% of patients [47]. In the canine model of AF, miR-29b was found to target Collagen-1A1 (COL1A1), Collagen-3A1 (COL3A1) and fibrilin [30], which are the prime markers of tissue remodeling. Moreover, the serum level of miR-29b is reduced in patients with AF by 54%, strongly implicating its contribution towards this ailment. The pro-inflammatory factors like Transforming Growth Factor-ß (TGF-ß) [48] and Nuclear Factor-?B (NF-?B) are reported to be the upstream regulators of miR-29 [49].
miR-26, miR-328 and miR-499: Role in regulation of ion channels
The miR-26 family (miR-26a-1, miR-26a-2 and miR-26b) can increase susceptibility to AF by altering the inward rectifier potassium current (IK1). IK1 is the major potassium current of myocardial repolarization, which partially regulate cell excitability and APD. The lower expression of miR-26 in patients with AF and murine atrial tissue induces KIR2.1 and KCNJ2 expression and shortens APD [29], suggesting that the downregulation of miRNA-26 promotes the formation of AF. MiR-26 activates T cell nuclear factor. An elevated T cell nuclear factor in atrial tissue, in turn, reduces the expression of miR-26, leading to the increase in inward rectifier current.
MiR-328 is involved in a variety of physiological and pathological processes of tissue organization, such as the formation of senile dementia [50], primary biliary cirrhosis [51] and chronic bladder pain syndrome [52]. AF is a characterized by abnormal excitability via myocardial calcium overload (e.g. the outflow rate of calcium ions), potentiating the development and maintenance of AF [53]. It has been reported that miR-328 plays an essential role in development of AF [54]. Through microarray analysis, a recent study has shown a 3.5 fold increase in the miR-328 level in eight week samples from a right atrial tachyarrhythmia canine model compared with control. It has also been shown that overexpression of miR-328 increases the susceptibility of animals towards AF, whereas blocking its expression through chemicals or gene knockdown reduces it. In-silico analysis has revealed that calcium channel regulatory genes, such as CACNA1C and CACNB1 (L-type calcium channels a1c and ß1 subunit, respectively), are the targets of miR-328 [36]. MiR-328-mediated inhibition of the L-type calcium channels results in shortening of the action potential duration enhancing the risk of AF development [55]. Furthermore, the expression of miR-328 was found to have a positive correlation with left atrial diameter, suggesting that miR-328 plays a central role in atrial electrical activity.
Mir-499 has been recently shown to target KCNN3 which encodes the small-conductance Ca2+-activated K+ channel 3 (SK3) and potentially contribute to the electrical remodeling event in AF [37].
miR-1 and miR-133: role in regulation of Ca2+ cycling
Mir-1 and miR-133 are two major muscle-specific miRNAs whose deregulation has been associated with the extrasystolic spontaneous Ca2+ release from sarcoplasmic reticulum and the generation of arrythmogenesis after depolarization [22,56]. Increased occurrence of spontaneous Ca2+ release at the molecular level was found to be due the dissociation of Ryanodine Receptors (RyR2s) with the Protein Phosphatase (PP2A) resulting in increased phosphorylation of RyR2s. In canine model of heart failure, high levels of miR-1 and miR-133 were found to be correlated with the down regulation of the regulatory and catalytic subunits (B56a and B56ß) of PP2A and the subsequent increase in Calcium/Calmodulin-dependent protein kinase II (CaMKII) mediated RyR2 hyperphosphorylation resulting in Ca2+ release [57].
ROLE OF MIRNAS IN THE MAINTENANCE OF AF
miR-21 and miR-590: Role in Collagen Deposition
MiR-590 has recently been correlated with AF due to its role in reducing collagen production and deposition, which is a significant source of atrial fibrosis. In nicotine induced canine models, collagen content was found to be significantly increased, coincident with an increase in TGF-ß/TGF-ßRII and a decrease in miR-590 levels [33].
miR-155 and miR-19: Role in activation of renin-angiotensin-aldosterone system
It has been shown that in the newborn mouse cardiomyocyte, the high level of miR-19b significantly reduces Ang II-induced CTGF expression, decreases the accumulation of atrial ECM, and prevents atrial fibrosis [26].
miR-133 and miR-590: Role in regulation of myogenesis and atrial remodeling
miR-30 and miR-208: Role in myocardial hypertrophy/atrial fibrosis
MiR-208 is mainly expressed in cardiac tissue and this family consists of miR-208a and miR-208b encoded by the intron of a cardiac muscle Myosin heavy chain gene (Myh6) and ß-cardiac muscle Myosin heavy chain gene (Myh7), respectively [65]. Thyroid Hormone-Associated Protein 1 (THRAP1) and myostatin, both of which are negative regulators of muscle hypertrophy, are reported targets of miR-208 family. Transgenic expression of miR-208 in murine heart has proven hypertrophic to the cardiac tissue due to decreased expression of THRAP1 and myostatin and is sufficient to induce arrhythmias [35].
SUMMARY AND PERSPECTIVE: MIRNA IN AF
AF is a common clinical arrhythmia, but the mechanisms are not yet fully understood. MiRNAs assist in regulation, generation, development and maintenance of AF via modulations in ion channels, receptors and extracellular matrix proteins. Some miRNAs implicated in AF have potential for use as biomarkers and drug targets [66]. For example, serum miR-21 and miR-29 can be used as a noninvasive method for detecting AF and inhibition of miR-21 may prevent myocardial fibrosis to reduce the risk of AF. However, our current knowledge about miRNA and its contribution towards AF is still limited and therefore, learning more about miRNAs will provide new insights into their mechanisms of action and help us in determining effective treatment targets [67]. Data from different studies show that some miRNAs are causative while others have protective effect for AF depending upon their targets. For example, some are pro-fibrotic while others are anti-fibrotic. Similarly, some are pro-arrythmatic whereas others are anti-arrythmatic. Nevertheless, we can identify the deregulated ones to be associated with the disease development and/or maintainance and use them as biomarkers, at least, for that ailment. Future research in this direction is needed, including gathering of larger data sets from animal models and in-depth analysis of the role of miRNAs in the pathogenesis of AF. The definitive function of a particular miRNA can however be evaluated by the knockdown studies, after which the therapeutic potential could be assessed. The functional analysis through miRNA overexpression and their knockdown through anti-miRNA oligos can provide insight about their role in the pathogenesis and/or maintenance of a particular disease. This will help in the development of clinical trials to test miRNA-based therapeutic drugs. In case of AF there is substantial evidence regarding the role of miRNAs in the development and maintenance of the disease. Thus, the beneficial effect of using miRNA for AF seems to hold a lot of promise for future treatments and therapies.
ACKNOWLEDGEMENTS
This work was supported by grants from National Natural Science Foundation of China (81272880), the Shanghai Committee of Science and Technology (124119b1300), Shanghai Municipal Health Bureau (2012-186) and a start-up fund of research from Jinshan Hospital (2012-2) to GX.
CONFLICT OF INTEREST STATEMENT
The authors have nothing to disclose.
REFERENCES
- Grönefeld GC, Lilienthal J, Kuck KH, Hohnloser SH; Pharmacological Intervention in Atrial Fibrillation (PIAF) Study investigators (2003) Impact of rate versus rhythm control on quality of life in patients with persistent atrial fibrillation. Results from a prospective randomized study. Eur Heart J 24: 1430-1436.
- Chien KL, Su TC, Hsu HC, Chang WT, Chen PC, et al. (2010) Atrial fibrillation prevalence, incidence and risk of stroke and all-cause death among Chinese. Int J Cardiol 139: 173-180.
- Chou CC, Nihei M, Zhou S, Tan A, Kawase A, et al. (2005) Intracellular calcium dynamics and anisotropic reentry in isolated canine pulmonary veins and left atrium. Circulation 111: 2889-2897.
- Nattel S (2002) New ideas about atrial fibrillation 50 years on. Nature 415: 219-226.
- Nattel S, Harada M (2014) Atrial remodeling and atrial fibrillation: recent advances and translational perspectives. J Am Coll Cardiol 63: 2335-2345.
- Nattel S, Burstein B, Dobrev D (2008) Atrial remodeling and atrial fibrillation: mechanisms and implications. Circ Arrhythm Electrophysiol 1: 62-73.
- Van Wagoner DR, Nattel S (2008) Insights into mechanisms linking cardiac hypertrophy and atrial fibrosis: evidence for a role of histone deacetylase in atrial fibrillation pathophysiology and therapy. J Mol Cell Cardiol 45: 707-708.
- Guo H, Ingolia NT, Weissman JS, Bartel DP (2010) Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466: 835-840.
- Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281-297.
- Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136: 215-233.
- Nilsen TW (2007) Mechanisms of microRNA-mediated gene regulation in animal cells. Trends Genet 23: 243-249.
- Hashimoto Y, Akiyama Y, Yuasa Y (2013) Multiple-to-multiple relationships between microRNAs and target genes in gastric cancer. PLoS One 8: 62589.
- Cai X, Hagedorn CH, Cullen BR (2004) Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 10: 1957-1966.
- Lee Y, Ahn C, Han J, Choi H, Kim J, et al. (2003) The nuclear RNase III Drosha initiates microRNA processing. Nature 425: 415-419.
- Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ (2004) Processing of primary microRNAs by the Microprocessor complex. Nature 432: 231-235.
- Yi R, Qin Y, Macara IG, Cullen BR (2003) Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev 17: 3011-3016.
- Gregory RI, Yan KP, Amuthan G, Chendrimada T, Doratotaj B, et al. (2004) The Microprocessor complex mediates the genesis of microRNAs. Nature 432: 235-240.
- Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, et al. (2000) The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403: 901-906.
- Urbich C, Kuehbacher A, Dimmeler S (2008) Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc Res 79: 581-588.
- Jakob P1, Landmesser U (2012) Role of microRNAs in stem/progenitor cells and cardiovascular repair. Cardiovasc Res 93: 614-622.
- Lionetti V, Ventura C (2013) Regenerative medicine approach to repair the failing heart. Vascul Pharmacol 58: 159-163.
- Kim GH (2013) MicroRNA regulation of cardiac conduction and arrhythmias. Transl Res 161: 381-392.
- Xiao J, Liang D, Zhang Y, Liu Y, Zhang H, et al. (2011) MicroRNA expression signature in atrial fibrillation with mitral stenosis. Physiol Genomics 43: 655-664.
- Liu H, Chen GX, Liang MY, Qin H, Rong J, et al. (2014) Atrial fibrillation alters the microRNA expression profiles of the left atria of patients with mitral stenosis. BMC Cardiovasc Disord 14: 10.
- Belevych AE, Sansom SE, Terentyeva R, Ho HT, Nishijima Y, et al. (2011) MicroRNA-1 and -133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RyR2 complex. PLoS One 6: e28324.
- Gao S, Liu TW, Wang Z, Jiao ZY, Cai J, et al. (2014) Downregulation of microRNA-19b contributes to angiotensin II-induced overexpression of connective tissue growth factor in cardiomyocytes. Cardiology 127: 114-120.
- Charles NJ, Huebert RC, Lee S, Adhikari N, Polster S, et al. (2010) Targeted Sprouty1 overexpression in cardiac myocytes does not alter myocardial remodeling or function. Mol Cell Biochem 342: 57-62.
- Thum T, Gross C, Fiedler J, Fischer T, Kissler S, et al. (2008) MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 456: 980-984.
- Luo X, Pan Z, Shan H, Xiao J, Sun X, et al. (2013) MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. J Clin Invest 123: 1939-1951.
- Dawson K, Wakili R, Ordög B, Clauss S, Chen Y, et al. (2013) MicroRNA29: a mechanistic contributor and potential biomarker in atrial fibrillation. Circulation 127: 1466-1475, 1475e1-28.
- Pan W, Zhong Y, Cheng C, Liu B, Wang L, et al. (2013) MiR-30-regulated autophagy mediates angiotensin II-induced myocardial hypertrophy. PLoS One 8: 53950.
- Feng Y, Niu LL, Wei W, Zhang WY, Li XY, et al. (2013) A feedback circuit between miR-133 and the ERK1/2 pathway involving an exquisite mechanism for regulating myoblast proliferation and differentiation. Cell Death Dis 4: e934.
- Shan H, Zhang Y, Lu Y, Zhang Y, Pan Z, et al. (2009) Downregulation of miR-133 and miR-590 contributes to nicotine-induced atrial remodelling in canines. Cardiovasc Res 83: 465-472.
- Martin MM, Buckenberger JA, Jiang J, Malana GE, Nuovo GJ, et al. (2007) The human angiotensin II type 1 receptor +1166 A/C polymorphism attenuates microRNA-155 binding. J Biol Chem 282: 24262-24269.
- Callis TE, Pandya K, Seok HY, Tang RH, Tatsuguchi M, et al. (2009) MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J Clin Invest 119: 2772-2786.
- Bodi I, Mikala G, Koch SE, Akhter SA, Schwartz A (2005) The L-type calcium channel in the heart: the beat goes on. J Clin Invest 115: 3306-3317.
- Ling TY, Wang XL, Chai Q, Lau TW, Koestler CM, et al. (2013) Regulation of the SK3 channel by microRNA-499--potential role in atrial fibrillation. Heart Rhythm 10: 1001-1009.
- Alegret JM, Aragonès G, Elosua R, Beltrán-Debón R, Hernández-Aguilera A, et al. (2013) The relevance of the association between inflammation and atrial fibrillation. Eur J Clin Invest 43: 324-331.
- Farmakis D, Parissis J, Filippatos G3 (2014) Insights into onco-cardiology: atrial fibrillation in cancer. J Am Coll Cardiol 63: 945-953.
- Goette A, Honeycutt C, Langberg JJ (1996) Electrical remodeling in atrial fibrillation. Time course and mechanisms. Circulation 94: 2968-2974.
- Roderburg C, Urban GW, Bettermann K, Vucur M, Zimmermann H, et al. (2011) Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology 53: 209-218.
- Cushing L, Kuang PP, Qian J, Shao F, Wu J, et al. (2011) miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am J Respir Cell Mol Biol 45: 287-294.
- Boon RA, Seeger T, Heydt S, Fischer A, Hergenreider E, et al. (2011) MicroRNA-29 in aortic dilation: implications for aneurysm formation. Circ Res 109: 1115-1119.
- Emilsson L, Smith JG, West J, Melander O, Ludvigsson JF (2011) Increased risk of atrial fibrillation in patients with coeliac disease: a nationwide cohort study. Eur Heart J 32: 2430-2437.
- Ahlehoff O, Gislason GH, Jørgensen CH, Lindhardsen J, Charlot M, et al. (2012) Psoriasis and risk of atrial fibrillation and ischaemic stroke: a Danish Nationwide Cohort Study. Eur Heart J 33: 2054-2064.
- Li H, Scherlag BJ, Kem DC, Zillner C, Male S, et al. (2013) Atrial tachycardia provoked in the presence of activating autoantibodies to β2-adrenergic receptor in the rabbit. Heart Rhythm 10: 436-441.
- Frustaci A, Chimenti C, Bellocci F, Morgante E, Russo MA, et al. (1997) Histological substrate of atrial biopsies in patients with lone atrial fibrillation. Circulation 96: 1180-1184.
- Zhang Y, Huang XR, Wei LH, Chung AC, Yu CM, et al. (2014) miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-β/Smad3 signaling. Mol Ther 22: 974-985.
- Mott JL, Kurita S, Cazanave SC, Bronk SF, Werneburg NW, et al. (2010) Transcriptional suppression of mir-29b-1/mir-29a promoter by c-Myc, hedgehog, and NF-kappaB. J Cell Biochem 110: 1155-1164.
- Boissonneault V, Plante I, Rivest S, Provost P (2009) MicroRNA-298 and microRNA-328 regulate expression of mouse beta-amyloid precursor protein-converting enzyme 1. J Biol Chem 284: 1971-1981.
- Padgett KA, Lan RY, Leung PC, Lleo A, Dawson K, et al. (2009) Primary biliary cirrhosis is associated with altered hepatic microRNA expression. J Autoimmun 32: 246-253.
- Sanchez Freire V, Burkhard FC, Kessler TM, Kuhn A, Draeger A, et al. (2010) MicroRNAs may mediate the down-regulation of neurokinin-1 receptor in chronic bladder pain syndrome. Am J Pathol 176: 288-303.
- Mancarella S, Yue Y, Karnabi E, Qu Y, El-Sherif N, et al. (2008) Impaired Ca2+ homeostasis is associated with atrial fibrillation in the alpha1D L-type Ca2+ channel KO mouse. Am J Physiol Heart Circ Physiol 295: H2017-2024.
- Lu Y, Zhang Y, Wang N, Pan Z, Gao X, et al. (2010) MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation 122: 2378-2387.
- Sood S, Chelu MG, van Oort RJ, Skapura D, Santonastasi M, et al. (2008) Intracellular calcium leak due to FKBP12.6 deficiency in mice facilitates the inducibility of atrial fibrillation. Heart Rhythm 5: 1047-1054.
- Terentyev D, Belevych AE, Terentyeva R, Martin MM, Malana GE, et al. (2009) miR-1 overexpression enhances Ca(2+) release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit B56alpha and causing CaMKII-dependent hyperphosphorylation of RyR2. Circ Res 104: 514-521.
- Nishijima Y, Feldman DS, Bonagura JD, Ozkanlar Y, Jenkins PJ, et al. (2005) Canine nonischemic left ventricular dysfunction: a model of chronic human cardiomyopathy. J Card Fail 11: 638-644.
- Adam O, Löhfelm B, Thum T, Gupta SK, Puhl SL, et al. (2012) Role of miR-21 in the pathogenesis of atrial fibrosis. Basic Res Cardiol 107: 278.
- Roy S, Khanna S, Hussain SR, Biswas S, Azad A, et al. (2009) MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc Res 82: 21-29.
- Goudis CA, Kallergis EM, Vardas PE (2012) Extracellular matrix alterations in the atria: insights into the mechanisms and perpetuation of atrial fibrillation. Europace 14: 623-630.
- Adam O, Theobald K, Lavall D, Grube M, Kroemer HK, et al. (2011) Increased lysyl oxidase expression and collagen cross-linking during atrial fibrillation. J Mol Cell Cardiol 50: 678-685.
- Nickenig G, Harrison DG (2002) The AT(1)-type angiotensin receptor in oxidative stress and atherogenesis: Part II: AT(1) receptor regulation. Circulation 105: 530-536.
- Li H, Li S, Yu B, Liu S (2012) Expression of miR-133 and miR-30 in chronic atrial fibrillation in canines. Mol Med Rep 5: 1457-1460.
- Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, et al. (2009) miR-133 and miR-30 regulate connective tissue growth factor: implications for a role of microRNAs in myocardial matrix remodeling. Circ Res 104: 170-178.
- van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, et al. (2007) Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 316: 575-579.
- Zhao Z, Liu T, Wang X, Li G (2013) MicroRNAs as novel antiarrhythmic targets for atrial fibrillation. Int J Cardiol 168: 135-137.
- Shi KH, Tao H, Yang JJ, Wu JX, Xu SS, et al. (2013) Role of microRNAs in atrial fibrillation: new insights and perspectives. Cell Signal 25: 2079-2084.
Citation: Peng F, Gong H, Nadeem L, Zhang L, Xu G (2014) Microrna Associated With Generation, Development, and Maintenance of Atrial Fibrillation. J Cardiol Stud Res 1: 003.
Copyright: © 2014 Fei Peng, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.