Mitochondria-Endoplasmic Reticulum Cross Talk in Premature Ovarian Insufficiency
*Corresponding Author(s):
Mona SharmaDepartment Of Reproductive Biology, AIIMS, New Delhi, India
Email:dr.mona18sharma@gmail.com
Abstract
Ovarian function is integral to women’s reproductive health. Mitochondria, recognized as the cellular powerhouses, are pivotal in sustaining cellular activities by generating ATP and regulating various processes such as cell division, redox reactions and apoptosis. All these processes are important events of folliculogenesis. Therefore, mitochondrial function is linked to cellular energy homeostasis during folliculogenesis. Mitochondria function in close association with the endoplasmic reticulum both at structural and molecular levels thereby regulating cellular harmony. With infertility emerging as a global health concern, understanding the role of mitochondria and its cross talk in fertility becomes crucial. This manuscript focuses on mitochondrial-endoplasmic reticulum crosstalk and its influence on ovarian folliculogenesis and its clinical implication in pathophysiology of premature ovarian insufficiency.
Keywords
Endoplasmic reticulum; Mitochondria; Premature ovarian insufficiency
Introduction
Ovarian function is integral to fertility and relies primarily on mitochondrial beta-oxidation for cellular function during folliculogenesis. Infertility has become a global health issue, with female infertility contributing to 37% of infertile couples [1]. The female factors contributing to infertility is estimated to be 15% in India [2]. Infertility arising due to ovarian dysfunction also known as Premature Ovarian Insufficiency (POI), constitutes roughly 1% of cases of infertility in females. During the mammalian reproductive lifetime, ovarian follicles mature through the course of folliculogenesis, in which a good quality oocyte is selected for ovulation. As a feature of the folliculogenesis process, a few follicles are disposed of by follicular atresia, chiefly by apoptosis. It has been shown that there are other known ways of programmed ovarian cell death apart from apoptosis, such as autophagy, paraptosis and necroptosis [3-5].
The excessive rate of follicular atresia is usually the cause of premature cessation of ovarian function, as in POI. Other causes of aberrant folliculogenesis causing female infertility include Polycystic Ovarian Syndrome (PCOS), where there is an increased number of immature follicles found. Multiple biomarkers have been studied for diagnosis of PCOS [6-8]. Mitochondrial activity has been explored in PCOS, and therefore, it is vital to study mitochondrial dynamics in the regulation of folliculogenesis [9]. Mitochondrial beta oxidation is essential for folliculogenesis, and the Mitochondrial Fatty Acid Shuttle System (MFAS) is a prerequisite for beta oxidation. Mitochondria function in close association with the Endoplasmic Reticulum (ER). Mitochondrial dysfunction has been shown to cause ER stress, which leads to cellular dysfunction. Therefore, mitochondrial-ER crosstalk is vital to understand folliculogenesis and ovarian function.
- Premature Ovarian Insufficiency
Folliculogenesis requires energy, which is derived mainly from mitochondrial beta oxidation [10]. Premature Ovarian Insufficiency (POI), also known as early menopause or premature menopause, is the cessation of ovarian function before 40 years of age [11]. POI occurs when the ovaries lose their germinative and hormonal functions because of the decline in ovarian follicles prior to the physiological age of menopause [12]. POI is defined by the loss of ovarian activity before the age of 40 years. Clinically, POI is diagnosed by menstrual disturbances (amenorrhea or oligomenorrhea) with raised gonadotropins (FSH level > 25 IU/L) and low estradiol (< 10 pg/ml). Multiple etiologies have been associated with POI, such as genetic, autoimmune, iatrogenic, nutritional, and environmental factors, but the majority of POI cases remain idiopathic [13-16]. POI poses a significant challenge to women’s fertility and lifelong health. The condition exhibits remarkable heterogeneity, encompassing a broad spectrum of phenotypes and etiologies. Usually, POI is diagnosed at later stages, but the pathogenesis starts much earlier.
- Mitochondria and MFAS
Mitochondria are double membrane-bound organelles known as the powerhouses of the cell, as they produce energy in the form of ATP, and their proper functioning is vital for various cellular activities, including cell division, redox balance, calcium homeostasis, adaptive thermogenesis and cell death or apoptosis [17,18]. Mitochondria also play a role to play in human fertility [19]. Fatty acid metabolism via beta oxidation is vital for folliculogenesis. MFAS is a prerequisite for beta oxidation and consists of l-Carnitine (LC), Organic Carnitine Transporter 2 (OCTN2), Coenzyme A (CoA), Carnitine Palmitoyltransferase I (CPT1), Carnitine Palmitoyltransferase II (CPT2), and Carnitine-Acylcarnitine Translocase (CACT) [20]. Long-chain fatty acyl-CoAs can cross the inner mitochondrial membrane only via shuttle proteins. Acyl-CoA molecules get conjugated to carnitine by CPT1. Acylcarnitines need CACT to be transported across the inner mitochondrial membrane. Free acyl-CoAs are released into the mitochondrial matrix by CPT2 and the free carnitine is then transported back to the cytoplasm (Figure 1).
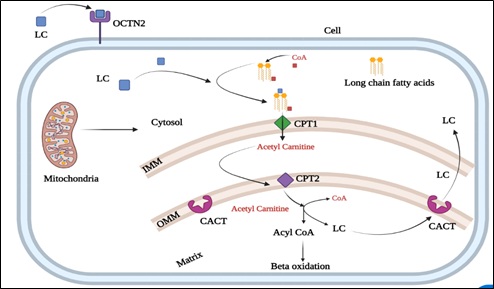 Figure1: Components of mitochondrial shuttle system.
Figure1: Components of mitochondrial shuttle system.
LC: L-carnitine; OCTN2: Organic carnitine transporter 2; CoA: coenzyme A; CPT1: Carnitine palmitoyltransferase I; CPT2: Carnitine palmitoyltransferase II; CACT: Carnitine-acylcarnitine translocase; IMM: Inner mitochondrial membrane; OMM: Outer mitochondrial membrane
- Mitochondria: Role in folliculogenesis
Folliculogenesis and oogenesis are well-coordinated events where the oocyte and surrounding granulosa cells influence each other's development [21]. During folliculogenesis, oocyte maturation passes through meiotic arrest and resumption phases, in which meiosis I is completed, and a secondary oocyte is formed that again gets arrested in metaphase II just before ovulation [22]. Mitochondrial proteins are needed during oocyte maturation and many in vitro studies have shown the role of LC and CPT1 in oocyte maturation [23-25]. It has been shown that the number of mitochondria increases during oogenesis and the maturation of oocytes [26]. The activity and distribution of mitochondria represent a critical point for oocyte maturation [27]. Previous literature has highlighted the importance of lipid metabolism in maintaining good oocyte quality [24,25]. Some in vitro maturation studies have indicated that LC supplementation increases the rate of beta-oxidation, fertilization and blastocyst development [28,29]. CPT1 is also critical for meiotic resumption in mice oocytes [24].
Oocyte maturation is linked to cumulus cells, which ensure metabolic substrates in the form of ATP [30]. Higher levels of ATP in cumulus-enclosed oocytes were observed, as cumulus cells provide metabolic support for oocytes [31]. Similarly, inhibition of beta-oxidation during in vitro maturation of bovine and mouse oocytes impairs blastocyst development [24,25]. CPT2 gene mutation has been linked to infertility and therefore has implications for in vitro fertilization [31]. CPT1 deficiency aggravates pressure overload-induced cardiac hypertrophy caused by lipotoxicity [32]. These data suggest that downregulation of MFAS leads to lipotoxicity, which may be detrimental for cell function. All these studies suggest that mitochondria and MFAS are critical for folliculogenesis and oocyte maturation.
- Mitochondria and endoplasmic reticulum crosstalk
ER and mitochondria are considered the cell's main source and sink of calcium, respectively. Mitochondria and ER interact with each other via specialized membranes known as Mitochondrial-Associated Membranes (MAMs). These mitochondrial membranes are associated with various important cellular functions, such as lipid synthesis, Ca2+ signaling, and the control of mitochondrial biogenesis and intracellular trafficking [33]. Inositol 1,4,5-Trisphosphate Receptor (IP3R) interacts with mitochondrial Voltage-Dependent Anion Channel 1 (VDAC1) at the Outer Mitochondrial Membrane (OMM) and is connected via the molecular chaperone Glucose Regulator 75 (GRP75), thereby regulating Ca2+ uptake by the OMM (Figure 2).
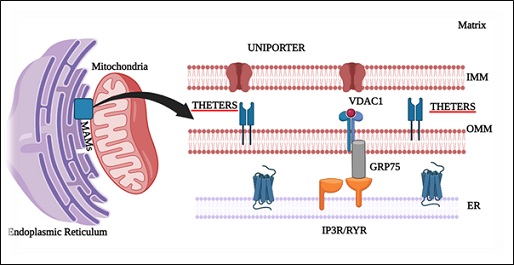 Figure 2: Endoplasmic reticulum and Mitochondria crosstalk via mitochondrial associated membranes (MAMs).
Figure 2: Endoplasmic reticulum and Mitochondria crosstalk via mitochondrial associated membranes (MAMs).
ER: Endoplasmic reticulum; IMM: Inner mitochondrial membrane; OMM: Outer mitochondrial membrane; VDAC1: Voltage-dependent anion-selective channel 1; GRP75: glucose-regulated protein 75; IP3R: inositol 1,4,5-trisphosphate receptor; RYR: Ryanodine receptors
The ATP synthesized via mitochondria is utilized by the Sarcoplasmic/Endoplasmic Reticulum Ca2+-ATPase (SERCA) channels on the ER membrane, which control the uptake of calcium in the ER lumen (Figure 3). Decreased mitochondrial ATP reduces calcium uptake into the ER, leading to by activating ER stress sensors [34]. Thus, decreased beta oxidation can lead to ER stress.
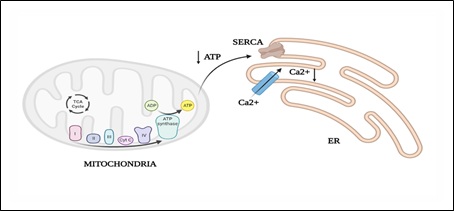 Figure 3: Mitochondrial ATP utilization by endoplasmic reticulum by SERCA channels.
Figure 3: Mitochondrial ATP utilization by endoplasmic reticulum by SERCA channels.
ER: Endoplasmic reticulum; Ca2+: calcium; SERCA: sarcoplasmic/endoplasmic reticulum Ca2+-ATPase
- Unfolded protein response and ER stress
ER function is essential for most cellular activities and survival. Abnormal ER function causes the accumulation and aggregation of unfolded proteins. Transmembrane receptors on the ER detect and initiate the Unfolded Protein Response (UPR) to bring back ER function. Should stress endure, or if the adaptive mechanism falters, apoptotic cell demise ensues. ER has been chiefly acknowledged as the hub for producing and correctly folding secreted, membrane-bound, and certain organelle-directed proteins. Several factors are required for ideal protein folding, including ATP, Ca2+, and an oxidizing environment to allow disulfide-bond formation [35]. As a result of this special oxidizing environment, the ER is highly sensitive to stresses that disturb cellular energy levels, the redox state, or Ca2+ concentration.
Protein folding capacity reduces as a result of cellular stress of the ER, resulting in the accumulation and aggregation of unfolded proteins, thereby causing ER stress. Protein aggregation is toxic to cells in numerous pathophysiological conditions such as ischemia, neurodegenerative diseases, and diabetes [36]. The orchestration of UPR primarily hinges on three ER transmembrane receptors: Pancreatic ER Kinase (PKR)-like ER Kinase (PERK), Activating Transcription Factor 6 (ATF6) and Inositol-Requiring Enzyme 1 (IRE1). In normal conditions when the cell is in the resting stage, all these three ER stress receptors are kept in an inactive state through their association with the ER chaperone, GRP78.
In this way, at whatever point there is an accumulation of unfolded proteins, GRP78 separates from the three receptors, prompting the enactment of UPR, which is a pro-survival response. UPR at first attempts to reduce the accumulation of unfolded proteins and reestablish normal ER function [37]. In case protein aggregation is persistent, the pro-survival signalling switches to pro-apoptotic. Molecular mechanisms behind this switching over are still needs to be explored. Upon aggregation of unfolded proteins, GRP78 dissociates from the three ER stress receptors, PERK, ATF6 and IRE1, allowing their activation.
There is series of activation of these receptors, with PERK being the first, followed by ATF6, and the last one to get initiated is IRE1. Activated PERK phosphorylates eukaryotic initiation factor 2α (eIF2α), which blocks protein synthesis. This is followed by translation of ATF4 by the eIF2α-independent translation pathway. ATF4 translocate to the nucleus and induces gene transcription to restore ER homeostasis. Another transcription factor ATF6 translocate from the ER to the Golgi apparatus and gets activated by proteolysis. Active ATF6 mediates the expression of ER chaperones and another transcription factor, X Box-Binding Protein 1 (XBP1). XBP1 undergoes mRNA splicing for its activation, which is carried out by IRE1. Spliced XBP1 Protein (sXBP1) then enters to the nucleus and leading to the transcription of chaperones, co-chaperones, and the PERK-inhibitor P58IPK. All these events ultimately try to normalise ER function by further inhibiting the buildup of proteins, increasing the folding capacity, and initiating the degradation of protein aggregates [38] (Figures 4 & 5).
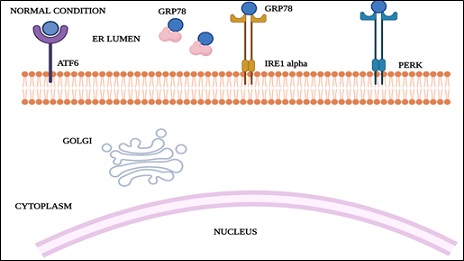 Figure 4: Endoplasmic reticulum during normal cell physiology.
Figure 4: Endoplasmic reticulum during normal cell physiology.
GRP78: glucose-regulated protein78; ATF6: activating transcription factor 6; IRE1 alpha: inositol-requiring enzyme 1; PERK: pancreatic ER kinase (PKR)-like ER kinase.
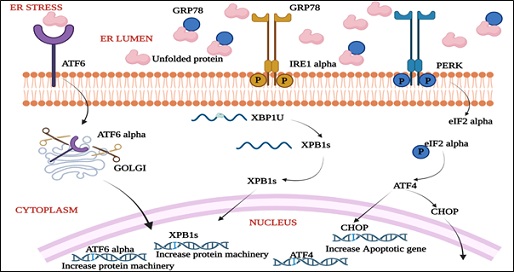 Figure 5: Unfolded Protein Response (UPR) signaling during stress conditions in ER.
Figure 5: Unfolded Protein Response (UPR) signaling during stress conditions in ER.
GRP78: glucose-regulated protein78; ATF6: activating transcription factor 6; IRE1 alpha: inositol-requiring enzyme 1;PERK: pancreatic ER kinase (PKR)-like ER kinase; CHOP: CCAAT-enhancer-binding protein homologous protein; XBP1: X-Box-Binding Protein; ATF4: activating transcription factor 4
- ER stress and apoptosis
During normal conditions, the pro-apoptotic proteins Bax and Bak (Bax/Bak) are kept in inactive by interaction with BCL2 present on mitochondrial as well as ER membranes, whereas Bim (BH3) binds to cytoskeletal dynein proteins and is inhibited. Severe and continous ER stress activates c-Jun N-terminal kinase (JNK) and CHOP protein (initiation phase). This eliminates the anti-apoptotic effect of BCL2. CHOP blocks BCL2 expression, whereas JNK phosphorylates BCL2. JNK phosphorylates Bim, thereby releasing it from the cytoskeleton and causing its activation (commitment phase). These changes enact Bax and Bak, sending signals from ER to the mitochondria that lead to the execution of cell demise (execution stage) [38] (Figure 6). Dysfunction of MFAS leads to the accumulation of fatty acids and lipotoxicity that can cause ER stress via the initation of ER stress sensors (Figure 7) [39].
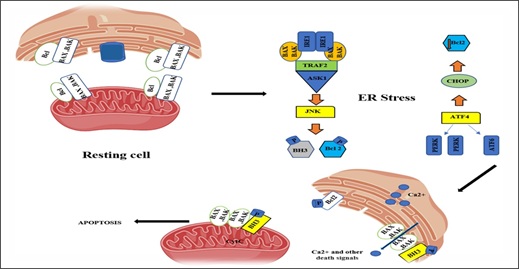 Figure 6: Endoplasmic reticulum stress leading to apoptosis.
Figure 6: Endoplasmic reticulum stress leading to apoptosis.
ATF4, ATF 6: activating transcription factor 4, 6; PERK, pancreatic ER kinase (PKR)-like ER kinase; TRAF2, TNF-receptor-associated factor 2; ASK1: Apoptosis signal regulating kinase 1
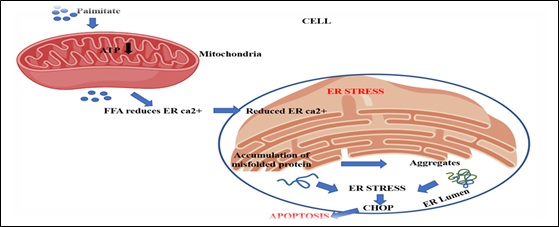 Figure 7: The figure illustrates the process of free fatty acids (Palmitate) accumulation in mitochondria, leading to ER stress and apoptosis.
Figure 7: The figure illustrates the process of free fatty acids (Palmitate) accumulation in mitochondria, leading to ER stress and apoptosis.
- ER stress and follicular atresia
The intrafollicular environment, rich in excess lipids, induces ER stress and impairs oocyte nuclear maturation. During folliculogenesis, UPR is observed in phases of follicular growth, maturation and atresia. Morphological ER disturbances and persistent ER stress lead to follicular atresia [40]. An exaggerated rate of follicular atresia is one of the major causes of POI, and the ER stress-induced apoptosis of granulosa cells and oocytes together lead to follicular atresia.
Future Directions
Aberrant function of MFAS can affect beta oxidation. It can be hypothesized that decreased beta-oxidation by causing decreased ATP production, lipotoxicity and ER Stress leads to abnormal follicular development or excessive follicular atresia leading to premature ovarian insufficiency.
The given hypothesis can be the basis of future studies where the effect of MFAS and ER Stress can be studied in understanding the pathophysiology of POI. The experimental animal model of POI can be created by using MFAS inhibitors, and its effect on ER Stress markers and ovarian morphology can be assessed. POI human cases can be studied for genetic aberrations also for biochemical disturbances in ER stress markers, MFAS protein expression, and apoptotic markers.
Conclusion
Normal folliculogenesis and oocyte maturation are integral to female reproductive health. The maintenance of a functional set of maternally derived mitochondria and functional crosstalk with the ER is pivotal for reproductive function and embryonic development. Mitochondrial-ER crosstalk is vital for cellular homeostasis and, consequently, for folliculogenesis. Therefore, studying the role of this inter-organelle communication in the pathophysiology of POI is crucial and equally intriguing.
Conflict of Interests
The author has no conflict of interest with any other parties.
Acknowledgment
None.
References
- Unuane D, Tournaye H, Velkeniers B, Poppe K (2011) Endocrine disorders & female infertility. Best Pract Res Clin Endocrinol Metab 25: 861-873.
- Rafique S, Sterling EW, Nelson LM (2012) A new approach to primary ovarian insufficiency. Obstet Gynecol Clin North Am 39: 567-586.
- Sharma M, Kumar R, Dhingra R (2014) Apoptosis: Researching the clinical implications. JIMSA 27: 161-162.
- Sharma M, Kumar R, Dhingra R (2012) Apoptosis: Understanding of the signaling pathways. IJBMR 3: 2029--2031.
- Sharma M, Kumar R, Dhingra R (2012) Apoptosis: Searching for the detection techniques. IJBMR 3: 2172-2175.
- Halder A, Kumar H, Sharma M, Jain M, Kalsi AK, et al. (2023) Serum anti-Müllerian hormone: A potential biomarker for polycystic ovary syndrome. Indian J Med Res 158: 397-406.
- Kumar H, Halder A, Sharma M, Jain M, Kalsi AK (2022) Dihydrotestosterone- A Potential Biomarker of Hyperandrogenaemia in Polycystic Ovary Syndrome: A Case-control Study from North India. JCDR 16: 09-14.
- Halder A, Kumar H, Sharma P, Jain M, Sharma M (2022) Polycystic Ovairan Syndrome (PCOS): An overview and our experience. J Endocrinol Reprod 26: 127-152.
- Zhang J , Bao Y, Zhou X, Zheng L (2019) Polycystic ovary syndrome and mitochondrial dysfunction. Reprod Biol Endocrino 17: 67.
- Dunning KR, Akison LK, Russell DL, Norman RJ, Robker RL (2011) Increased beta-oxidation and improved oocyte developmental competence in response to l-carnitine during ovarian in vitro follicle development in mice. Biol Reprod 85: 548-555.
- Sharma M, Sihag K, Halder A, Sharma JB (2023) Premature ovarian insufficiency in adolescents. J Indian Obstetrics Gynecology 13.
- Jankowska K (2017) Premature ovarian failure. Prz Menopauzalny 16: 51-56.
- Rishi I, Halder A, Sharma JB, Jain M, Sharma M (2020) Single Strand Conformation Polymorphism and Sequencing of HS6ST2 Gene in Patients of Idiopathic Premature Ovarian Failure. JCDR 14: 01-08.
- Rana R, Sharma M, Halder A (2022) New Insights into the Mechanism of Pathogenesis of Fragile X-Associated Premature Ovarian Failure. EC Gynaecology 11: 38-43.
- Sharma M, Halder A (2021) Understanding Basic Concepts of Premature Ovarian Failure. EC Gynaecolog 10: 25-36.
- Sharma M (2021) Nutrimiromics: A Possible Answer for Premature Ovarian Failure. Biochem Physiol 10: 292.
- St John JC, Facucho-Oliveira J, Jiang Y, Kelly R, Salah R (2010) Mitochondrial DNA transmission, replication and inheritance: a journey from the gamete through the embryo and into offspring and embryonic stem cells. Hum Reprod Update 16: 488-509.
- Dumollard R, Duchen M, Carroll J (2007) The role of mitochondrial function in the oocyte and embryo. Curr Top Dev Biol 77: 21-49.
- Barbhuiya PN, Gogoi A, Ahmed G, Mahanta R (2016) Prevalence of Mitochondrial DNA Nucleotide Substitution Mutations in Male Infertile Cases of Northeast India. Journal of Infertility and Reproductive Biology 4(1): 11-21.
- Sharma M (2018) Mitochondrial Fatty Acid Transport System and its Relevance to Ovarian Function. J Infertil Reprod Biol 6: 1-3.
- Sharma M (2022) Oogenesis and folliculogenesis. Manual of assisted reproductive technologies and laboratory sciences. In: Devi MG, Talwar P, Mehta J, Agarwal A (eds.). Evangel Publishing 1: 13-17.
- Sihag K, Sharma M (2021) Oocyte Metaphase Arrest and Release: Triggers and Pathways. J Infertil Reprod Biol 9: 59-64.
- Rana R, Halder A, Gupta S, Sharma M (2022) An in vitro analysis of L-Carnitine mediated rescue of TNF-α induced apoptosis in mice oocytes. Int J Clin Biochem Res 9: 218-223.
- Rana R, Sharma M (2018) A Timeline Morphological Study of TNF-α Induced Changes in Mice Oocytes. J Infertil Reprod Biol 6: 16-18.
- Downs SM, Mosey JL, Klinger J (2009) Fatty acid oxidation and meiotic resumption in mouse oocytes. Mol Reprod Dev 76: 844-853.
- Ramalho-Santos J, Varum S, Amaral S, Mota PC, Sousa AP, et al. (2009) Mitochondrial functionality in reproduction: From gonads and gametes to embryos and embryonic stem cells. Hum Reprod Update 15: 553-572.
- Nazmara Z, Salehnia M, HosseinKhani S (2014) Mitochondrial distribution and ATP content of vitrified, in vitro matured mouse oocytes. Avicenna J Med Biotechnol 6: 210-217.
- Carrillo-González DF, Maldonado-Estrada JG (2020) L-carnitine supplementation in culture media improves the pregnancy rate of in vitro produced embryos with sexed semen from Bos taurus indicus cows. Trop Anim Health Prod 52: 2559-2565.
- Moawad AR, Salama A, Badr MR, Fathi M (2021) Beneficial effects of L-carnitine supplementation during IVM of canine oocytes on their nuclear maturation and development in vitro. Animals (Basel) 11: 581.
- Dunning KR, Russell DL, Robker RL (2014) Lipids and oocyte developmental competence: The role of fatty acids and β-oxid Reproduction 148: 15-27.
- Hull ML, Nemeth D, Hague WM, Wilkinson C, Liebelt J, et al. (2009) Mitochondrial fatty acid transport enzyme deficiency--implications for in vitro Fertil Steril 91: 2732.
- Lin P, Yang Y, Li X, Chen F, Cui C, et al. (2012) Endoplasmic reticulum stress is involved in granulosa cell apoptosis during follicular atresia in goat ovaries. Mol Reprod Dev 79: 423-432.
- Hayashi T, Rizzuto R, Hajnoczky G, Su TP (2009) MAM: More than just a housekeeper. Trends Cell Biol 19: 81-88.
- Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R (2008) Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27: 6407-6418.
- Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, et al. (2005) PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J 24: 717-729.
- Kaufman RJ (2002) Orchestrating the unfolded protein response in health and disease. J Clin Invest 110: 1389-1398.
- Schröder M, Kaufman RJ (2005) ER stress and the unfolded protein response. Mutat Res 569: 29-63.
- Szegezdi E, Logue SE, Gorman AM, Samali A (2006) Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO reports 7: 880-885.
- Yoon H, Shaw JL, Haigis MC, Greka A (2021) Lipid metabolism in sickness and in health: Emerging regulators of lipotoxicity. Mol Cell 81: 3708-3730.
- Yang X, Wu LL, Chura LR, Liang X, Lane M, et al. (2012) Exposure to lipid-rich follicular fluid is associated with endoplasmic reticulum stress and impaired oocyte maturation in cumulus-oocyte complexes. Fertil Steril 97: 1438-1443.
Citation: Sihag K, Sharma M, Halder A, Sharma JB, Ara S (2024) Mitochondria-Endoplasmic Reticulum Cross Talk in Premature Ovarian Insufficiency. J Reprod Med Gynecol Obstet 9: 165.
Copyright: © 2024 Kajal Sihag, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.