Molecular Chaperones and Ubiquitin Proteasome System in Tumor Biogenesis: An Overview
*Corresponding Author(s):
Rashmi K AmbastaMolecular Neuroscience And Functional Genomics Laboratory, Delhi Technological University, Delhi, India
Tel:+91 9818898638,
Email:rashmiambasta@dce.edu; rashmiambasta@gmail.com
Abstract
The hallmark feature in cancer is uncontrolled cell-division and altered protein expression. Currently, cancer is one of the most detrimental diseases encountered by a large population across the globe. However, an absolute treatment strategy has still not been achieved by the researchers. Further, molecular mechanism and therapeutic to combat this lethal disease is a baffling issue. Molecular chaperones and ubiquitin proteasome system is mainly responsible for the maintenance of protein homeostasis and thus playing a crucial role in the cancer pathophysiology. Molecular chaperones are a superfamily of proteins which expressions are triggered under physiological, pharmacological and environmental insults and playing a protective role for cell survival. However, beyond a threshold of protection, molecular chaperones are unable to provide proper shape of non functional proteins that accumulate unwanted protein the cellular milieu. In order to get rid off these accumulated proteins ubiquitin proteasome system comes into action where an E3 ligase, specific enzymes of ubiquitination system, play a decisive role in the turnover of many essential regulatory proteins involved in cancer. It also mediates numerous functions, for instance, cell death, cell growth and DNA repair. Since, both molecular chaperones and E3 ligases have been involved in the progression of cancers it is necessary to understand and implement the role of these two molecules to use as diagnostic markers to treat cancer. Herein, we have comprehensively discussed the functional role of molecular chaperones, their differential protein expressions and a possible correction mechanism in cancer. Furthermore, comprehensive information has been documented regarding E3 ligases and their associated role in cancer that may be used as potential diagnostic biomarker for the treatment of various cancers.
Keywords
INTRODUCTION
The progression of cancer is so silent that it is hard to diagnose at the earlier stages, and it is an equally challenging task for the clinicians to treat this disease at early stage due to many unknown mechanisms with sites and tissues specificity issues. Cancer cells are immensely proliferatives, incursive, and metastatic in nature. The pathological hallmark in cancer is uncontrolled cellular proliferation and altered protein expression where hindrance in chaperoning machinery, and UPS is one of the major reasons. Molecular chaperones and ubiquitin E3 ligases are ubiquitous class of proteins that play an essential role in the conformational quality control of the proteins through interacting with various misfolded proteins, stabilizing and remodeling a wide range of non-native polypeptides [1,2]. Even though constitutively expressed under balanced growth conditions, many chaperones are upregulated upon exposure of heat shock or other insults that constitutively increases cellular protein level and for that reason, it is also known as stress or Heat Shock Proteins (HSPs) [3]. Molecular chaperones act together on unfolded or partially folded protein subunits, for instance, nascent chains emerging from the ribosome and with extended chains being translocated across sub-cellular membranes [4]. In addition, some chaperones are non-specific, and they easily interact with a broad range of protein subunits, while others are restricted to specific targets. They often require ATP binding/hydrolysis for their proper functions [4]. Moreover, they also stabilize the non-native conformation for correct folding of protein subunits [5] and not associate with native proteins nor do they form part of the final folded structures. HSPs were first identified as a group of proteins, which are brought about by heat shock and other kind of sources, including physical and chemical stressors in a broad range of species [6]. The HSPs have been afterwards described as molecular chaperones, which share common property of altering the structures and interactions of other proteins [7].
Importantly, molecular chaperone directs that the HSPs frequently interact in a stoichiometric manner with their substrates and also require high intracellular levels of proteins [6]. This is the reason why HSP28, HSP40, HSP70 and HSP110 genes have developed a highly effective mechanism for their synthesis during stress with transcriptional activation, efficient messenger RNA (mRNA) stabilization and their preferential translation [8] and boosts to become dominantly expressed proteins under such stress scenario [9]. Heat shock factor (hsf) family is the main regulatory machineries for HSP gene transcription, which ensure proper transcriptional activation and equally steep switch-off mechanism after recovery [10]. The hsf gene family comprises heat shock transcription factor 1 (hsf1), acts as central co-ordinator of heat shock stress response. Besides this, heat shock transcription factor 2 (hsf2) and heat shock transcription factor 4 (hsf4) are also involved in stress response but less active with respect to hsf1 [11]. HSP transcription needs activated hsf1, which is itself over-expressed in cancer and play a crucial role in invasion and metastasis [12]. Further, the molecular mechanisms that connecting increased HSP and hsf1 expression to tumor development are not presently well understood. Since, HSPs are activated only under stress conditions in the normal cells; thus play a decisive role in both cellular homeostasis and the stress response [13]. To maintain the protein homeostasis, cells also encompass a large number of constitutively expressed HSPs which are present in a variety of multiprotein complexes together with many co-factors [8,14]. These HSPs comprise HSP10 and HSP60 multi complexes that direct protein folding; whereas, HSP70 and HSP90 containing complexes are implicated in both generic protein-folding pathways in specific connection with key regulatory proteins within the cell [15]. Most importantly, HSP90 plays a central role in forming complexes with a large number of transcription factors, cellular kinases and other molecules that in turn involved in the cell regulation process. Levels of the HSPs are raised in numerous tumor cells and HSP over-expression marks a poor prognosis in context with cell-survival and response to therapy in specific cancer types [16]. An elevated HSP expression in malignant cells plays a critical role in protection from spontaneous apoptosis associated with malignancy as well as apoptosis caused due to therapy. Further, this mechanism may underlie a highly complex role of HSPs in cancer development and reluctance to treatment [17].
Exposure of cells to stress condition, including oxidative stress, heat shock, heavy metals, inflammation, infection, tissue damage and mutant proteins associated with the genetic disease’s result into an inducible expression of HSPs that acts as molecular chaperones [18]. HSPs are commonly found in both prokaryotic and eukaryotic cells and play a pivotal role in the cellular processes [19]. Interestingly, mammalian HSPs are grouped into four major families according to their molecular mass, including HSP90, HSP70, HSP60 and small HSPs [20]. Under physiological stress such as hypoxia, HSPs palying an essential role in the cell protection [21]. Further, HSP27, HSP70 and HSP90 are considered as anti-apoptotic markers, since; they were able to bind to some pro-apoptotic molecules, including cytochrome c and Apaf [22]. In contrast, HSP10 and HSP60 were suggested as pro-apoptotic [23]. It was also revealed that HSP70 increases the tumorigenicity of cancer cells in rodent models and inhibits apoptosis [24] and therefore, increases the survival of cells exposed to a wide range of lethal stimuli. Molecular chaperones have also ubiquitin-dependent proteasome like activity; ensure that damaged and short-lived proteins are degraded efficiently, thereby restoring protein homeostasis and promoting cell-survival [25]. Nevertheless, the altered expression of molecular chaperones and E3 ligases has been noticed in various cancers, including breast, prostate, lungs, and gastric. On contrary, these machineries are also having an opposite effect since, in many instances, it has been involved in inhibiting disease progression. Therefore, this review underlies the potential role of molecular chaperones and well-known E3 ligases in cancer biology. Further, this review will also provide a promising therapeutic strategy for clinicians to design suitable drugs for treating cancers.
AN INTERLINK BETWEEN CHAPERONING MACHINERY AND CHAPERONOPATHIES
The term ‘chaperoning system’ is postulated in 2008 and notion behind this is consolidated protective machineries that have molecular chaperones, co-chaperones and co-factors of an organism [26]. Chaperonopathies is a pathological condition which comprises chaperones as etiological pathogenic factors while, chaperonotherapy is used as a medical term in which chaperones are used for the treatment of various diseases [27]. This scheme visualizes a physiological system integrated by chaperones, their functionally-related molecules and structures that are present in all tissues, organs and biological fluids. The ultimate fate of this physiological system is pivotal for the control and maintenance of protein homeostasis by correcting and providing functional conformation of proteins in all body fluids and tissues [27]. The chaperoning system also contributes their major input in some other processes, including, receptor assembly, antigen presentation and the formation of complexes with a variety of ligands and several other cellular functions [28]. Further, the term chaperonology, a scientific discipline has been identifiedwhich is dealing with the chaperoning system and includes the study of the genomic sequences of chaperone genes by virtue of applying chaperonomics concept. Chaperones are highly conserved proteins and assist other proteins for proper folding and re-folding [29]. Moreover, when the proteins are irreversibly misfolded, they are destined for degradation [30]. Additionally, these chaperone proteins are involved in regulating immune system, cell differentiation, cell-signaling, gene expression, DNA replication, apoptosis, cellular senescence and oncogenesis [31]. These chaperones are not present only in the tissues but also in fluids such as cell secretion products and have hormone-like activity because they act in locations other than those of their origin. A number of molecular chaperones are HSPs which including HSP110, HSP90, HSP70, HSP60 and so on. These proteins are induced under heat shock insult as well as by other physical and chemical stressors in a broad range of species [32]. Furthermore, these HSPs are encoded in genes that are inducible by stressors, but many others are not HSPs. These HSPs delineate their major input to protein homeostasis and also play other roles more or less unconnected to the maintenance of protein homeostasis. Earlier, HSP genes were categorized according to their molecular mass expressed in kilodalton (kDa) elaborated in table 1. However, recently guidelines for the nomenclature of the human HSP families have been changed. The main motto of this nomenclature system is based on gene symbols, which have been attributed or assigned by the HUGO Gene Nomenclature Committee (HGNC) and are used as the primary identifiers such as entrez gene and ensemble database [45].
| HSPS | Associated Chaperones | Molecular Weight (kDa) | Functions | References |
| HSP40 | dnaJ, Ydj1, Sis1, Hdj1 and Hdj2 | 35-54 | Associated with protein folding and refolding along with HSP70/HSC70. It enhances the rate of Adenosine Triphosphatease (ATP) activity and substrate release | [33,34] |
| HSP60 | groEL, HSP60, Cpn60 and HSP60 | 55-64 | Helps in proper folding of proteins and also prevents aggregation of denatured proteins in vitro. It may also facilitate protein degradation by the help of proteolytic systems and involves in macrophage activation possibly through Toll-like receptor | [35] |
| HSP70 |
DnaK, Kar2, Ssc1, HSC70, HSP70, BIP and Mhsp70 |
65-80 | Assists in proper folding of proteins and also contribute their potential role in protein-trafficking. It has also potential anti-apoptotic like properties. Further, it has been found to be Involved in cell-cycle progression and proliferation | [36,37] |
| HSP90 | HtpG, HSP83, HSP90 and GRP94 | 81-99 | Involves in signal transduction process and also assists proper folding and maintenance of proteins in in vitro and crucial for cell-cycle control | [38] |
| HSP100 | ClpA,B,C and HSP104 | 90 | Assists in the resolubilization of heat-inactivated proteins from insoluble aggregates and also plays pivotal role in stress tolerance | [35] |
| Small HSPs | lbp A and B, HSP27, A and B crystalline and HSP27 | 34 or less than 34 | Helps in heat inactivation and suppression of proteins in in vitro. It has also potential anti-apoptotic like properties | [39,40] |
| CCT | Hsp70 and TRiC | 55-63 | Cytosolic molecular chaperone that is involved in Protein folding | [41,42] |
| HSP10 | HSP60 | 10 | Pivotal for protein folding along with HSP60 and modulation of immune system | [43] |
| HSP 27 | HspB1 | 20-30 | Helps in the refolding of denatured proteins and have anti-apoptotic like properties | [44] |
Table 1: Heat shock proteins, associated chaperones and their functional significance.
DISCUSSION
Clinical presentation of a FMH depends, on the one hand, on the volume of the transfusion and, on the other hand, on the velocity with which it had occurred [5]. The prognosis also depends on the prompt diagnosis and intervention.
FMH can occur as an acute or chronic event. In the chronic presentation, the hemorrhage has been prolonged or repeated during pregnancy, anemia developed slowly, giving the fetus the opportunity to develop hemodynamic compensation. In this case, the diagnosis is often postnatal and these infants may manifest only pallor at birth with no other complications [5].
In opposition, in acute FMH, perinatal hypoxia and intrauterine death or severe anemia and hypoxia at birth can be present [5].
Neonatal anemia was the first manifestation of FMH in 35% of the reported cases. In severe ones, shock and circulatory failure may be present [6].
In our clinical report, it’s more likely to be a chronic transfusion given the rapid ability of the newborn to adapt to extra uterine life despite the hemoglobin value at birth. The absence of fetal movements noted by the mother was not due to the FMH but it was caused by the circular of the umbilical cord around the arms that made it impossible to the fetus to move.
Abdominal trauma and invasive techniques of prenatal diagnosis are related to FMH [1]. Physicians should consider alternative diagnosis to neonatal anemia such as isoimmune hemolytic anemia, congenital infections that result in bone marrow suppression (TORCH), sepsis, congenital erythrocyte defects and congenital hemoglobinopathies [6]. Clinical and laboratory evaluation of infection, Coombs test and viral serology should be performed.
In this case, the diagnosis of FMH was confirmed by the KB test. Pink fetal red blood cells are observed and counted in the mother’s peripheral blood smear because fetal hemoglobin is resistant to acid elution, leaving discolored maternal cells (patients with sickle cell anemia or hereditary persistence of fetal hemoglobin may lead to a false positive result and ABO incompatibility may produce a false negative result) [2].
Although the KB test is inexpensive and requires no special equipment, it lacks standardization and is imprecise [3]. Flow cytometry, based on the use of anti-fetal hemoglobin for detection of fetal cells with fetal hemoglobin, represents an improvement of KB test since is more specific and precise [7].
Although the prognosis of massive FMH is poor, it can be improved if physicians early recognize this condition. When the infant is near-term gestation, immediate cesarean delivery is indicated. If, on the other hand, the fetus is still preterm, in uterus transfusion can be considered and has been shown to be effective and improves the prognosis [6].
Long term outcome for infants affected by massive FMH is unfavorable with death or neurological dysfunction [6]. The prognosis is more directly related to initial hemoglobin value and clinical manifestations post-delivery than with the transfused volume of blood [8].
The case reported emerged from a pregnancy with no risk factors. Mother’s perception of decreased fetal movements, recognition of fetal distress on the CTG, immediate caesarean section and prompt hemodynamic and respiratory support to the newborn with early red blood cells transfusion contributed for this good outcome.
A lethal complication of chaperoning machinery is considered to be a morbid condition where molecular chaperone malfunction is an etiopathological issue, and fatal complications (chaperonopathies) may be categorized as familial or acquired. In familial complication, the abnormal function of a chaperone is due to gene mutation [46]. Further, familial complications comprising the diseases of nervous system such as motor neuropathies and ataxias, heart disease includes familial forms of dilated cardiomyopathies and other sporadic forms of congenital diseases. In acquired chaperonopathies mediated complications, the malfunction of a chaperone may be due to morbid post-translational modifications, including oxidation, glycation, phosphorylation and acetylation resulting into loss or gain of function. Among the acquired chaperonopathies mediated complications, diseases are basically related with aging and with dysregulation of the immune system [46]. These fatal conditions may happen in any age. However, genetic chaperonopathies have a fairly early onset, while acquired chaperonopathies become exposed in the elderly, occasionally in association with other diseases. It has also been revealed that few prominent groups of chaperone are represented by the ‘chaperonopathies by mistake’ in which a molecular chaperone may be normal, although takes part in a cascade which indulges disease progression rather than the disease reversal [47]. In such condition, the pathology can be manifested by two types of pathogenic cascades; first the pathway is usual and comprises the participation of one or more chaperones which develops into part of a morbid process; and second the cascade that includes chaperones is not part of a physiological process within the cell, although comes to be potent and functional in morbid cases. In both situations, the chaperones conduce to disease progression ‘by mistake’ [46]. In some occasions, autoimmune diseases can also be evoked by molecular chaperone acting as an auto-antigen ‘by mistake’ or due to molecular mimicry between human and bacterial HSP60 and other human proteins [48,49]. Interestingly, in some cases of neoplasms, chaperones can promote growth and proliferation of tumor cells [46]. Thus, molecular chaperone has both the beneficial and negative impact on the cells, as in normal condition it directs favourable impact on the cell, however, any mutation in its genes leads to morbid ambiance, which is also spelled as chaperonopathies which is a prominent notion as it directs to novel anti-cancer strategies.
HEAT SHOCK PROTEINS: AN INTERVENOR IN TUMOR BIOGENESIS
HSP27
HSP27 along with HSP70 also serves as an anti-apoptotic factor by binding and sequestering cytosolic cytochrome c and therefore, creates a blockade for APAF-1 assembly of the apoptosome complex [57-59]. Furthermore, the activation of ‘death’ receptors such as TNF and TNF receptor superfamily member 6 (FAS, CD95 and APO-1) propels the formation of a death inducing complex for the activation of pro-caspase-8 to activate caspases by inducing the release of cytochrome c [6,60]. The phosphorylation pattern of HSP27 in tumor cells has also appeared distinct and shown different characteristic features as compared to the phosphorylation pattern of HSP27 in untransformed cells. Subsequently, it has been proposed that the diversity of HSP27 phosphorylation isoforms can also signify the useful tumor markers. Moreover, when phosphorylated HSP27 interacts with Daxx protein, it interferes with the interaction between same protein Daxx and Fas thereby blocking the FAS-mediated apoptosis to prevent cell death [61,62].
HSP60
HSP70
In cancer tissues, HSP70 is expressed at high levels, and their expression has been routinely linked with poor prognosis of tumors. The precise role of HSP70 in the cancer physiology is remains unsettled. Though, in cancer cells high expression of HSP70 is thought to provide a survival advantage due to its potential to stall apoptosis and senescence [77,78]. HSP70 also acts as the co-chaperone for HSP90 because of its role in the substrate-loading phase of the HSP90, which is important to the stability and function of multiple oncoproteins [79]. Further, elevated expression of HSP70 has also been correlated with therapeutic resistance while, the detailed mechanisms about resistance is still unknown. Moreover, recent evidence suggests that reduced activation of NF-κB, ERK and JNK pathways may be responsible for its therapeutic resistance [80]. Pharmacological inhibition of HSP90 has been found to provoke a compensatory activation of HSP70. This might be since HSP70 is a highly protective protein, which may strongly reduce the cell death effect induced by HSP90 inhibition. Consequently, the potential remedial advantage of regulating HSP70 activity has become striking, particularly dual therapy against both HSP70 and HSP90 [81-84]. It also acts as an inhibitor to progress towards G1 and S phase of the cell-cycle in human tumor cells and can be used as an attractive biomarker for cancers [85].
HSP90
HSP90 is an imperative molecular-chaperone that modulates the stability and activity of numerous client proteins, for instance, HER2, BCL-ABL, SRC, EGFR, CRAF, AKT, BRAF, MET, VEGFR, FLT3, HIF-1α, androgen and estrogen receptor and telomerase. These proteins directly involve in malignancy and are essential for the transformed phenotype [89]. In fact, elevated expression of HSP90 has been observed in a variety of human malignancies, including lung, breast tumors and leukaemias. In the breast cancer, it was found to associate with various proteins critical in breast cancer progression, such as HER2 and c-SRC [50]. Moreover, it also stabilizes many oncogenic proteins in tumor cells. It can inhibit apoptosis through several interactions. For illustration, it can bind directly with Apaf-1 thereby blocking the assembly of apoptosome complex. However, anti-apoptotic action of HSP90 has been revealed by its association with proteins important for generating a survival signal in response to growth factor stimulators. For instance, HSP90 can bind with AKT1, an anti-apoptotic protein to prevent its inactivation and favoring cell-survival. Furthermore, over-expression of HSP90 has been implicated in resistance to senescence due to its pivotal role in telomerase stability [90,91].
HSP110
HSP110 gene is a favourable molecular forecaster in Colorectal Cancer (CRC). Its association with common clinicopathological information may be the decisive for diagnosis of the lymph node metastatic mediated cancer prognosis, identifying more aggressive tumors and in predicting the clinical outcome thus, might facilitate clinical oncologists in designing more aggressive remedies for subgroups of patients with colorectal cancer [96]. Mutant form of HSP110 in colorectal cancer showing microsatellite instability caused from an aberrantly spliced mRNA and lacking the HSP110 substrate-binding domain. This mutant was expressed at variable levels in almost all microsatellite instability CRC cell lines. Mutated HSP110 weakened both the normal cellular localization of HSP110 and its interaction with other HSPs, thus, opposing the chaperone activity and anti-apoptotic function of HSP110 in a dominant-negative manner. An over-expression of this mutated form of HSP110 resulted in the sensitization of cells to anti-cancer agents such as oxaliplatin and 5-fluorouracil, which are usually medically advised in the adjuvant treatment of people with CRC. HSP110 may thus, establish a crucial determining factor for both prognosis and medication response in CRC [97]. The signaling axis where HSPs mainly trigger their potential role has been elaborating in figure 1. Despite the involvement of molecular chaperones in cancer, the pertinent role of UPS is also needed to be explored to understand its significance in cancer biology.
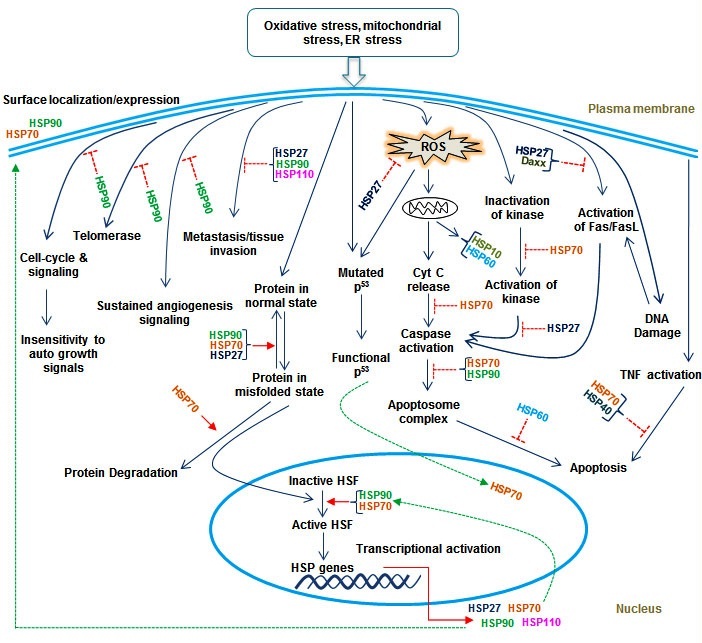
Figure 1: Pictorial representation of the common mechanism for the role of HSPs in various signaling targets.
Ubiquitin/Proteasome Pathway: A Central Player Of Protein Homeostasis Machineries
There are several types of malignancies, for example, breast, gastric, colon, renal and cervical cancer has been reported to be associated with altered UPS. Further, there are approximately thousand of E3 ligases in the human genome have been reported so far, that can be differentiated into three major groups based upon their domain structure and substrate recognition. The first group comprises N-end rule ubiquitin ligase that targets protein substrates allowing to destabilize N-terminal residues, including Arg, Lys, His (type, I) and Phe, Trp, Leu, Tyr and Ile (type II). The current example of protein degradation by the Ub-dependent N-end rule cascade is Drosophila inhibitor of apoptosis protein. The second type of E3 is HECT known as E6-associated protein, which function is related with oncoprotein E6, responsible for ubiquitin mediated degradation of p53. The third and largest type of E3 ligase is known as the RING family which carries a classic RING finger domain with a linear sequence of Cys-X-Cys-X-Cys-X-His-X-Cys/His-X-Cys-X-Cys-X-Cys, where X can be any amino acid. A RING finger domain attaches to two zinc atoms per molecule, where the first and third pairs of cysteine/histidine form the first binding site, and the second and fourth pairs of cysteine/histidine form the other binding site. Further, E3 ubiquitin ligases exist and proceed as a single peptide (such as Mdm2 and XIAP or as multiple component complexes, for instance, SCF). Through the covalent modification of a massive repertoire of cellular proteins with ubiquitin, E3 ligases control almost all aspects of eukaryotic cellular biological process and function. However, deregulated expression of E3 ligases leads to cancer progression, although over-expression of E3 ligases is associated with reduced pathogenesis of disease. Thus, E3 ligases which find out the specificity of protein substrates and themselves critical enzymes may provide potential cancer targets in addition to cancer biomarkers [100-104]. It also functions in tissue specific and targets specific manner with maximal therapeutic index with minimal toxicity and provides a potential cancer target in addition to cancer biomarkers [105]. This notion, therefore, makes E3 ligase a potential biomarker/forecaster for cancers.
E3 LIGASES: A POTENTIAL BIOMARKER FOR CANCER DIAGNOSIS
CUL4A
ARF-BP1
EFP
RNF115/BCA2
SKP2
BTRC1
WWP1
SIAH1
BRCA1
CHFR and CUL-5
FBXW7/CDC4
Parkin
CHIP/STUB1
| S.No. | E3 ligases | Associated functions | Principle phenotypes | References |
| 1 | AMFR/RNF45 | AMFR plays a critical role in metastasis through ubiquitination of the metastasis suppressor KAI1 | Prostate, liver and breast cancer | [148] |
| 2 | HACE1 | Acts as tumor suppressor. Moreover, abnormal methylation and Rac 1 signaling make it oncogenic | Wilms’ tumors, colorectal and breast cancer | [149] |
| 3 | PJA1/PRAJA | It plays an important role in downregulation of TGF-β signaling via ubiquitination of the SMAD4 adaptor protein ELF | Gastric cancer | [150] |
| 4 | Cbl-b | Cbl-b along with TAM receptors regulate cancer metastasis via natural killer cells | ------- | [151] |
| 5 | UBE3A/ E6-AP | It interacts with the E6 protein of the human papillomavirus types 16 and 18, and activates p53 for degradation | Breast cancer | [152] |
| 6 | HUWE1/MULE | It suppresses RAS-driven skin tumorigenesis in vivo by regulating c-MYC/MIZ1 stability | Skin cancer | [153] |
| 7 | RFWD2/COP1 | It ubiquitinates several proteins involved in the DNA damage response and apoptosis, including MTA1, p53 and FoxO1. Further, it also inactivates proto-oncogenes (ETV1, ETV4 and ETV5) | Prostate cancer | [154] |
| 8 | RNF5 | It involves in motility, stress response and regulates cancer progression through cytoskeleton alterations | Breast cancer | [155] |
| 9 | RNF13 | It contributes to a reduction in the colonization of metastatic tumor cells | Lung and pancreatic cancer | [156] |
| 10 | FBXO11 | It is associated with HIF-1α for its oncogenic effects. Further, it acts as a tumor suppressor and also to be a potential target for cancer therapy | Gynecological cancer | [157] |
| 11 | SPOP | It acts as tumor-suppressor and involved in signal-dependent proteolysis of the oncogenic co-activator SRC-3/AIB1 | Liver and breast cancer | [158] |
| 12 | SMURF2 | It promotes Epithelial-Mesenchymal Transition (EMT) in HPDE6c7 cells and its over-expression inhibits TGF-β-mediated EMT in the cells. Subsequently, it is also found to be involved with metastasis | Pancreatic and breast cancer | [159] |
| 13 | RNF31 | It stabilizes estrogen receptor α and modulates estrogen-stimulated cell proliferation | Breast cancer | [160] |
| 14 | RNF43 | RNF43 interacts with NEDL1 and modulates p53-mediated transcription | Colorectal cancer | [161] |
Table 2: Potential E3 ligases and their associated functionalities in cancers.
CONCLUSION
The main goal of cancer therapies relies on the researcher’s endowment to adjust actions to circumstances and has a clear projection pertaining to the aberrant mechanisms that ultimately determine the fate of the pathogenesis and henceforth degeneration through diverse processes. Despite gradual improvement in the field of cancer therapies, still the destined of such therapies hangs on morbid speculation and fragile anticipations. The diverse signaling axis offerings, an interesting opportunity to exploit in the pathology of cancers; furthermore, interrupting critical interactions of the pathways by using potential diagnostic marker such as, E3 ligases and molecular chaperones can solve the “targeted therapy crisis” problem in cancer biology. In doing so, HSPs are concerned with the pathogenesis and development of a range of human cancer types. They primarily participate in crucial mechanisms of tumor cells, including invasiveness, cell proliferation and neoangiogenesis, differentiation, metastasis, and immune system recognition. Since, HSPs are presently considered as biomarkers of carcinogenesis and their expression is correlated with the extent of differentiation and aggressiveness of certain tumors. Therefore, HSPs may also have been potential as forecasters of response to therapy. As well, their use as anti-cancer agents is growing, for instance, as inducers of anti-tumor immune responses. Importantly, E3 ligases have also shown the regulatory role in various cancers and lead to protect from a variety of cancers, conversely, it also acts as oncogenes. These E3 ligases have a significant role in motility, cytoskeleton alterations, cell growths, cell-death, DNA repairs and cell-cycle progression. This notion advocates new insights for anticancer therapy. Nevertheless, further research is needed in order to make drugs/compounds in cancer therapeutics a blatant reality.
ACKNOWLEDGEMENT
The authors would like to thank SERB/DST for funding the project. The authors would also like to thank the administration of DTU for encouragement and excellent infrastructure provided.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
- Vabulas RM, Raychaudhuri S, Hayer-Hartl M, Hartl FU (2010) Protein folding in the cytoplasm and the heat shock response. Cold Spring Harb Perspect Biol 2: 004390.
- Chen B, Retzlaff M, Roos T, Frydman J (2011) Cellular strategies of protein quality control. Cold Spring Harb Perspect Biol 3: 004374.
- Morano KA, Grant CM, Moye-Rowley WS (2012) The response to heat shock and oxidative stress in Saccharomyces cerevisiae. Genetics 190: 1157-1195.
- Hartl FU, Bracher A, Hayer-Hartl M (2011) Molecular chaperones in protein folding and proteostasis. Nature 475: 324-332.
- Saibil H (2013) Chaperone machines for protein folding, unfolding and disaggregation. Nat Rev Mol Cell Biol 14: 630-642.
- Martínez-Paz P, Morales M, Martín R, Martínez-Guitarte JL, Morcillo G (2014) Characterization of the small heat shock protein Hsp27 gene in Chironomus riparius (Diptera) and its expression profile in response to temperature changes and xenobiotic exposures. Cell Stress Chaperones 19: 529-540.
- Requena JM, Montalvo AM, Fraga J (2015) Molecular Chaperones of Leishmania: Central Players in Many Stress-Related and -Unrelated Physiological Processes BioMed Research International 2015: 301-326.
- Tang D, Khaleque MA, Jones EL, Theriault JR, Li C, et al. (2005) Expression of heat shock proteins and heat shock protein messenger ribonucleic acid in human prostate carcinoma in vitro and in tumors in vivo. Cell Stress Chaperones 10: 46-58.
- Zhao M, Tang D, Lechpammer S, Hoffman A, Asea A, et al. (2002) Double-stranded RNA-dependent protein kinase (pkr) is essential for thermotolerance, accumulation of HSP70, and stabilization of ARE-containing HSP70 mRNA during stress. J Biol Chem 277: 44539-44547.
- Calderwood SK, Xie Y, Wang X, Khaleque MA, Chou SD, et al. (2010) Signal Transduction Pathways Leading to Heat Shock Transcription. Sign Transduct Insights 2: 13-24.
- Akerfelt M, Morimoto RI, Sistonen L (2010) Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol 11: 545-555.
- Wang MH, Grossmann ME, Young CY (2004) Forced expression of heat-shock protein 70 increases the secretion of Hsp70 and provides protection against tumour growth. Br J Cancer 90: 926-931.
- Lindquist S, Craig EA (1988) The heat-shock proteins. Annu Rev Genet 22: 631-677.
- Buchner J (1999) Hsp90 & Co. - a holding for folding. Trends Biochem Sci 24: 136-141.
- Pratt WB, Toft DO (2003) Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood) 228: 111-133.
- van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AA, et al. (2002) A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 347: 1999-2009.
- Gyrd-Hansen M, Nylandsted J, Jäättelä M (2004) Heat shock protein 70 promotes cancer cell viability by safeguarding lysosomal integrity. Cell Cycle 3: 1484-1485.
- Akerfelt M, Morimoto RI, Sistonen L (2010) Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol 11: 545-555.
- Zügel U, Kaufmann SH (1999) Role of heat shock proteins in protection from and pathogenesis of infectious diseases. Clin Microbiol Rev 12: 19-39.
- Yang Z, Zhuang L, Szatmary P, Wen L, Sun H, et al. (2015) Upregulation of heat shock proteins (HSPA12A, HSP90B1, HSPA4, HSPA5 and HSPA6) in tumour tissues is associated with poor outcomes from HBV-related early-stage hepatocellular carcinoma. Int J Med Sci 12: 256-263.
- Fan GC (2012) Role of heat shock proteins in stem cell behavior. Prog Mol Biol Transl Sci 111: 305-322.
- Purandhar K, Jena PK, Prajapati B, Rajput P, Seshadri S (2014) Understanding the role of heat shock protein isoforms in male fertility, aging and apoptosis. World J Mens Health 32: 123-132.
- Shan YX, Liu TJ, Su HF, Samsamshariat A, Mestril R, et al. (2003) Hsp10 and Hsp60 modulate Bcl-2 family and mitochondria apoptosis signaling induced by doxorubicin in cardiac muscle cells. J Mol Cell Cardiol 35: 1135-1143.
- Rérole AL, Jego G, Garrido C (2011) Hsp70: anti-apoptotic and tumorigenic protein. Methods Mol Biol 787: 205-230.
- Shang F, Taylor A (2012) Roles for the ubiquitin-proteasome pathway in protein quality control and signaling in the retina: implications in the pathogenesis of age-related macular degeneration. Mol Aspects Med 33: 446-466.
- Tomala K, Korona R (2008) Molecular chaperones and selection against mutations. Biol Direct 3: 5.
- Cappello F, Marino Gammazza A, Palumbo Piccionello A, Campanella C, Pace A, et al. (2014) Hsp60 chaperonopathies and chaperonotherapy: targets and agents. Expert Opin Ther Targets 18: 185-208.
- Mayer MP, Bukau B (2005) Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci 62: 670-684.
- Kampinga HH, Craig EA (2010) The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol 11: 579-592.
- Stolz A, Wolf DH (2010) Endoplasmic reticulum associated protein degradation: a chaperone assisted journey to hell. Biochim Biophys Acta 1803: 694-705.
- Rappa F, Farina F, Zummo G, David S, Campanella C, et al. (2012) HSP-molecular chaperones in cancer biogenesis and tumor therapy: an overview. Anticancer Res 32: 5139-5150.
- Mahmood K, Jadoon S, Mahmood Q, Irshad M, Hussain J (2014) Synergistic effects of toxic elements on heat shock proteins. Biomed Res Int 2014: 564136.
- Qiu XB, Shao YM, Miao S, Wang L (2006) The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol Life Sci 63: 2560-2570.
- Lopez N, Aron R, Craig EA (2003) Specificity of class II Hsp40 Sis1 in maintenance of yeast prion [RNQ+]. Mol Biol Cell 14: 1172-1181.
- Parsell DA, Taulien J, Lindquist S (1993) The role of heat-shock proteins in thermotolerance. Philos Trans R Soc Lond B Biol Sci 339: 279-285.
- Mosser DD, Caron AW, Bourget L, Denis-Larose C, Massie B (1997) Role of the human heat shock protein hsp70 in protection against stress-induced apoptosis. Mol Cell Biol 17: 5317-5327.
- Buzzard KA, Giaccia AJ, Killender M, Anderson RL (1998) Heat shock protein 72 modulates pathways of stress-induced apoptosis. J Biol Chem 273: 17147-17153.
- Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, et al. (1997) Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell 89: 239-250.
- Richards EH, Hickey E, Weber L, Master JR (1996) Effect of overexpression of the small heat shock protein HSP27 on the heat and drug sensitivities of human testis tumor cells. Cancer Res 56: 2446-2451.
- Trautinger F, Kokesch C, Herbacek I, Knobler RM, Kindås-Mügge I (1997) Overexpression of the small heat shock protein, hsp27, confers resistance to hyperthermia, but not to oxidative stress and UV-induced cell death, in a stably transfected squamous cell carcinoma cell line. J Photochem Photobiol B 39: 90-95.
- Kubota H (2002) Function and regulation of cytosolic molecular chaperone CCT. Vitam Horm 65: 313-331.
- Melville MW, McClellan AJ, Meyer AS, Darveau A, Frydman J (2003) The Hsp70 and TRiC/CCT chaperone systems cooperate in vivo to assemble the von Hippel-Lindau tumor suppressor complex. Mol Cell Biol 23: 3141-3151.
- Höhfeld J, Hartl FU (1994) Role of the chaperonin cofactor Hsp10 in protein folding and sorting in yeast mitochondria. J Cell Biol 126: 305-315.
- Arrigo AP (2007) The cellular “networking” of mammalian Hsp27 and its functions in the control of protein folding, redox state and apoptosis. Adv Exp Med Biol 594: 14-26.
- Kampinga HH, Hageman J, Vos MJ, Kubota H, Tanguay RM, et al. (2009) Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 14: 105-111.
- Macario AJ, Conway de Macario E (2007) Chaperonopathies and chaperonotherapy. FEBS Lett 581: 3681-3688.
- Savvateeva-Popova E, Popov A, Grossman A, Nikitina E, Medvedeva A, et al. (2007) Pathogenic chaperone-like RNA induces congophilic aggregates and facilitates neurodegeneration in Drosophila. Cell Stress Chaperones 12: 9-19.
- Rodolico V, Tomasello G, Zerilli M, Martorana A, Pitruzzella A, et al. (2010) Hsp60 and Hsp10 increase in colon mucosa of Crohn’s disease and ulcerative colitis. Cell Stress Chaperones 15: 877-884.
- Gammazza AM, Bucchieri F, Grimaldi LM, Benigno A, de Macario EC, et al. (2012) The molecular anatomy of human HSP60 and its similarity with that of bacterial orthologs and acetylcholine receptor reveal a potential pathogenetic role of anti-chaperonin immunity in myasthenia gravis. Cell Mol Neurobiol 32: 943-947.
- Ciocca DR, Oesterreich S, Chamness GC, McGuire WL, Fuqua SA (1993) Biological and clinical implications of heat shock protein 27,000 (Hsp27): a review. J Natl Cancer Inst 85: 1558-1570.
- Jakob U, Gaestel M, Engel K, Buchner J (1993) Small heat shock proteins are molecular chaperones. J Biol Chem 268: 1517-1520.
- Garrido C, Brunet M, Didelot C, Zermati Y, Schmitt E, et al. (2006) Heat shock proteins 27 and 70: anti-apoptotic proteins with tumorigenic properties. Cell Cycle 5: 2592-2601.
- Myung JK, Afjehi-Sadat L, Felizardo-Cabatic M, Slavc I, Lubec G (2004) Expressional patterns of chaperones in ten human tumor cell lines. Proteome Sci 2: 8.
- Brunet Simioni M, De Thonel A, Hammann A, Joly AL, Bossis G, et al. (2009) Heat shock protein 27 is involved in SUMO-2/3 modification of heat shock factor 1 and thereby modulates the transcription factor activity. Oncogene 28: 3332-3344.
- Katsogiannou M, Andrieu C, Rocchi P (2014) Heat shock protein 27 phosphorylation state is associated with cancer progression. Front Genet 5: 346.
- Kim LS, Kim JH (2011) Heat shock protein as molecular targets for breast cancer therapeutics. J Breast Cancer 14: 167-174.
- Lee SJ, Choi SA, Lee KH, Chung HY, Kim TH, et al. (2001) Role of inducible heat shock protein 70 in radiation-induced cell death. Cell Stress Chaperones 6: 273-281.
- Zorzi E, Bonvini P (2011) Inducible hsp70 in the regulation of cancer cell survival: analysis of chaperone induction, expression and activity. Cancers (Basel) 3: 3921-3956.
- Kennedy D, Jäger R, Mosser DD, Samali A (2014) Regulation of apoptosis by heat shock proteins. IUBMB Life 66: 327-338.
- Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35: 495-516.
- Charette SJ, Lavoie JN, Lambert H, Landry J (2000) Inhibition of Daxx-mediated apoptosis by heat shock protein 27. Mol Cell Biol 20: 7602-7612.
- Ferns G, Shams S, Shafi S (2006) Heat shock protein 27: its potential role in vascular disease. Int J Exp Pathol 87: 253-274.
- Ghosh JC, Dohi T, Kang BH, Altieri DC (2008) Hsp60 regulation of tumor cell apoptosis. J Biol Chem 283: 5188-5194.
- Cappello F, David S, Rappa F, Bucchieri F, Marasà L, et al. (2005) The expression of HSP60 and HSP10 in large bowel carcinomas with lymph node metastase. BMC Cancer 5: 139.
- Ciocca DR, Calderwood SK (2005) Heat shock proteins in cancer: diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 10: 86-103.
- Korennykh A, Walter P (2012) Structural basis of the unfolded protein response. Annu Rev Cell Dev Biol 28: 251-277.
- Wang Q, He Z, Zhang J, Wang Y, Wang T, et al. (2005) Overexpression of endoplasmic reticulum molecular chaperone GRP94 and GRP78 in human lung cancer tissues and its significance. Cancer Detect Prev 29: 544-551.
- Pratt WB (1998) The hsp90-based chaperone system: involvement in signal transduction from a variety of hormone and growth factor receptors. Proc Soc Exp Biol Med 217: 420-434.
- Schaffitzel E, Rüdiger S, Bukau B, Deuerling E (2001) Functional dissection of trigger factor and DnaK: interactions with nascent polypeptides and thermally denatured proteins. Biol Chem 382: 1235-1243.
- Young JC, Barral JM, Ulrich Hartl F (2003) More than folding: localized functions of cytosolic chaperones. Trends Biochem Sci 28: 541-547.
- Bercovich B, Stancovski I, Mayer A, Blumenfeld N, Laszlo A, et al. (1997) Ubiquitin-dependent degradation of certain protein substrates in vitro requires the molecular chaperone Hsc70. J Biol Chem 272: 9002-9010.
- Murphy ME (2013) The HSP70 family and cancer. Carcinogenesis 34: 1181-1188.
- Zourlidou A, Payne Smith MD, Latchman DS (2004) HSP27 but not HSP70 has a potent protective effect against alpha-synuclein-induced cell death in mammalian neuronal cells. J Neurochem 88: 1439-1448.
- Li H, Liu L, Xing D, Chen WR (2010) Inhibition of the JNK/Bim pathway by Hsp70 prevents Bax activation in UV-induced apoptosis. FEBS Lett 584: 4672-4678.
- Beere HM (2005) Death versus survival: functional interaction between the apoptotic and stress-inducible heat shock protein pathways. J Clin Invest 115: 2633-2639.
- Juhasz K, Lipp AM, Nimmervoll B, Sonnleitner A, Hesse J, et al. (2013) The complex function of hsp70 in metastatic cancer. Cancers (Basel) 6: 42-66.
- Powers MV, Jones K, Barillari C, Westwood I, van Montfort RL, et al. (2010) Targeting HSP70: the second potentially druggable heat shock protein and molecular chaperone? Cell Cycle 9: 1542-1550.
- Gabai VL, Meriin AB, Yaglom JA, Volloch VZ, Sherman MY (1998) Role of Hsp70 in regulation of stress-kinase JNK: implications in apoptosis and aging. FEBS Lett 438: 1-4.
- Li J, Soroka J, Buchner J (2012) The Hsp90 chaperone machinery: conformational dynamics and regulation by co-chaperones. Biochim Biophys Acta 1823: 624-635.
- Nahleh Z, Tfayli A, Najm A, El Sayed A, Nahle Z (2012) Heat shock proteins in cancer: targeting the ‘chaperones’. Future Med Chem 4: 927-935.
- Patury S, Miyata Y, Gestwicki JE (2009) Pharmacological targeting of the Hsp70 chaperone. Curr Top Med Chem 9: 1337-1351.
- Gabai VL, Budagova KR, Sherman MY (2005) Increased expression of the major heat shock protein Hsp72 in human prostate carcinoma cells is dispensable for their viability but confers resistance to a variety of anticancer agents. Oncogene 24: 3328-3338.
- Bottoni P, Giardina B, Scatena R (2009) Proteomic profiling of heat shock proteins: An emerging molecular approach with direct pathophysiological and clinical implications. Proteomics Clin Appl 3: 636-653.
- Powers MV, Clarke PA, Workman P (2008) Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell 14: 250-262.
- Neganova I, Lako M (2008) G1 to S phase cell cycle transition in somatic and embryonic stem cells. J Anat 213: 30-44.
- Sreedhar AS, Kalmár E, Csermely P, Shen YF (2004) Hsp90 isoforms: functions, expression and clinical importance. FEBS Lett 562: 11-15.
- Dias S, Shmelkov SV, Lam G, Rafii S (2002) VEGF(165) promotes survival of leukemic cells by Hsp90-mediated induction of Bcl-2 expression and apoptosis inhibition. Blood 99: 2532-2540.
- Whitesell L, Lindquist SL (2005) HSP90 and the chaperoning of cancer. Nat Rev Cancer 5: 761-772.
- Zhao R, Kakihara Y, Gribun A, Huen J, Yang G, et al. (2008) Molecular chaperone Hsp90 stabilizes Pih1/Nop17 to maintain R2TP complex activity that regulates snoRNA accumulation. J Cell Biol 180: 563-578.
- Lanneau D, de Thonel A, Maurel S, Didelot C, Garrido C (2007) Apoptosis versus cell differentiation: role of heat shock proteins HSP90, HSP70 and HSP27. Prion 1: 53-60.
- Ciocca DR, Arrigo AP, Calderwood SK (2013) Heat shock proteins and heat shock factor 1 in carcinogenesis and tumor development: an update. Arch Toxicol 87: 19-48.
- Easton DP, Kaneko Y, Subjeck JR (2000) The hsp110 and Grp1 70 stress proteins: newly recognized relatives of the Hsp70s. Cell Stress Chaperones 5: 276-290.
- Romanucci M, Bastow T, Della Salda L (2008) Heat shock proteins in animal neoplasms and human tumours--a comparison. Cell Stress Chaperones 13: 253-262.
- Hosaka S, Nakatsura T, Tsukamoto H, Hatayama T, Baba H, et al. (2006) Synthetic small interfering RNA targeting heat shock protein 105 induces apoptosis of various cancer cells both in vitro and in vivo. Cancer Sci 97: 623-632.
- Slaby O, Sobkova K, Svoboda M, Garajova I, Fabian P, et al. (2009) Significant overexpression of Hsp110 gene during colorectal cancer progression. Oncol Rep 21: 1235-1241.
- Kim JH, Kang GH (2014) Molecular and prognostic heterogeneity of microsatellite-unstable colorectal cancer. World J Gastroenterol 20: 4230-4243.
- Dorard C, de Thonel A, Collura A, Marisa L, Svrcek M, et al. (2011) Expression of a mutant HSP110 sensitizes colorectal cancer cells to chemotherapy and improves disease prognosis. Nat Med 17: 1283-1289.
- Ciechanover A (1998) The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J 17: 7151-7160.
- Pickart CM (2001) Ubiquitin enters the new millennium. Mol Cell 8: 499-504.
- Varshavsky A (2003) The N-end rule and regulation of apoptosis. Nat Cell Biol 5: 373-376.
- Ditzel M, Wilson R, Tenev T, Zachariou A, Paul A, et al. (2003) Degradation of DIAP1 by the N-end rule pathway is essential for regulating apoptosis. Nat Cell Biol 5: 467-473.
- Scheffner M, Huibregtse JM, Vierstra RD, Howley PM (1993) The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75: 495-505.
- Freemont PS (2000) RING for destruction? Curr Biol 10: R84-87.
- Nakayama KI, Nakayama K (2006) Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer 6: 369-381.
- Sun Y (2006) E3 ubiquitin ligases as cancer targets and biomarkers. Neoplasia 8: 645-654.
- Sharma P, Nag A (2014) CUL4A ubiquitin ligase: a promising drug target for cancer and other human diseases. Open Biol 4: 130217.
- Zhao Y, Sun Y (2013) Cullin-RING Ligases as attractive anti-cancer targets. Curr Pharm Des 19: 3215-3225.
- Hannah J, Zhou PB (2013) The CUL4A ubiquitin ligase is a potential therapeutic target in skin cancer and other malignancies. Chin J Cancer 32: 478-82.
- Lee J, Zhou P (2012) Pathogenic Role of the CRL4 Ubiquitin Ligase in Human Disease. Front Oncol 2: 21.
- Korzeniewski N, Zheng L, Cuevas R, Parry J, Chatterjee P, et al. (2009) Cullin 1 functions as a centrosomal suppressor of centriole multiplication by regulating polo-like kinase 4 protein levels. Cancer Res 69: 6668-6675.
- Chen C, Seth AK, Aplin AE (2006) Genetic and expression aberrations of E3 ubiquitin ligases in human breast cancer. Mol Cancer Res 4: 695-707.
- Zhao KW, Sikriwal D, Dong X, Guo P, Sun X, et al. (2011) Oestrogen causes degradation of KLF5 by inducing the E3 ubiquitin ligase EFP in ER-positive breast cancer cells. Biochem J 437: 323-333.
- Urano T, Saito T, Tsukui T, Fujita M, Hosoi T, et al. (2002) Efp targets 14-3-3 sigma for proteolysis and promotes breast tumour growth. Nature 417: 871-875.
- Horie K, Urano T, Inoue S (2003) Efp as a new molecular target for breast cancer therapy. Anticancer Drugs 14: 1-2.
- Burger AM, Gao Y, Amemiya Y, Kahn HJ, Kitching R, et al. (2005) A novel RING-type ubiquitin ligase breast cancer-associated gene 2 correlates with outcome in invasive breast cancer. Cancer Res 65: 10401-10412.
- Wang Z, Nie Z, Chen W, Zhou Z, Kong Q, et al. (2013) RNF115/BCA2 E3 ubiquitin ligase promotes breast cancer cell proliferation through targeting p21Waf1/Cip1 for ubiquitin-mediated degradation. Neoplasia 15: 1028-1035.
- Hao JJ, Shi ZZ, Zhao ZX, Zhang Y, Gong T, et al. (2012) Characterization of genetic rearrangements in esophageal squamous carcinoma cell lines by a combination of M-FISH and array-CGH: further confirmation of some split genomic regions in primary tumors. BMC Cancer 12: 367.
- Tedesco D, Lukas J, Reed SI (2002) The pRb-related protein p130 is regulated by phosphorylation-dependent proteolysis via the protein-ubiquitin ligase SCF(Skp2). Genes Dev 16: 2946-2957.
- Kudo Y, Guardavaccaro D, Santamaria PG, Koyama-Nasu R, Latres E, et al. (2004) Role of F-box protein betaTrcp1 in mammary gland development and tumorigenesis. Mol Cell Biol 24: 8184-8194.
- Wan M, Huang J, Jhala NC, Tytler EM, Yang L, et al. (2005) SCF(β-TrCP1) controls Smad4 protein stability in pancreatic cancer cells. Am J Pathol 166: 1379-1392.
- Busino L, Donzelli M, Chiesa M, Guardavaccaro D, Ganoth D, et al. (2003) Degradation of Cdc25A by β-TrCP during S phase and in response to DNA damage. Nature 426: 87-91.
- Chen C, Zhou Z, Ross JS, Zhou W, Dong JT (2007) The amplified WWP1 gene is a potential molecular target in breast cancer. Int J Cancer 121: 80-87.
- Yeung B, Ho KC, Yang X (2013) WWP1 E3 ligase targets LATS1 for ubiquitin-mediated degradation in breast cancer cells. PLoS One 8: e61027.
- Carrano AC, Dillin A2, Hunter T3 (2014) A Krüppel-like factor downstream of the E3 ligase WWP-1 mediates dietary-restriction-induced longevity in Caenorhabditis elegans. Nat Commun 5: 3772.
- Yang B, Kumar S (2010) Nedd4 and Nedd4-2: closely related ubiquitin-protein ligases with distinct physiological functions. Cell Death Differ 17: 68-77.
- Cho HS, Kelly JD, Hayami S, Toyokawa G, Takawa M, et al. (2011) Enhanced expression of EHMT2 is involved in the proliferation of cancer cells through negative regulation of SIAH1. Neoplasia 13: 676-684.
- Bruzzoni-Giovanelli H, Fernandez P, Veiga L, Podgorniak MP, Powell DJ, et al. (2010) Distinct expression patterns of the E3 ligase SIAH-1 and its partner Kid/KIF22 in normal tissues and in the breast tumoral processes. J Exp Clin Cancer Res 29: 10.
- Susini L, Passer BJ, Amzallag-Elbaz N, Juven-Gershon T, Prieur S, et al. (2001) Siah-1 binds and regulates the function of Numb. Proc Natl Acad Sci USA 98: 15067-15072.
- Germani A, Prabel A, Mourah S, Podgorniak MP, Di Carlo A, et al. (2003) SIAH-1 interacts with CtIP and promotes its degradation by the proteasome pathway. Oncogene 22: 8845-8851.
- Knauer SK, Mahendrarajah N2, Roos WP2, Krämer OH3 (2015) The inducible E3 ubiquitin ligases SIAH1 and SIAH2 perform critical roles in breast and prostate cancers. Cytokine Growth Factor Rev 26: 405-413.
- Silver DP, Livingston DM (2012) Mechanisms of BRCA1 tumor suppression. Cancer Discov 2: 679-684.
- Starita LM, Machida Y, Sankaran S, Elias JE, Griffin K, et al. (2004) BRCA1-dependent ubiquitination of gamma-tubulin regulates centrosome number. Mol Cell Biol 24: 8457-8466.
- Dong Y, Hakimi MA, Chen X, Kumaraswamy E, Cooch NS, et al. (2003) Regulation of BRCC, a holoenzyme complex containing BRCA1 and BRCA2, by a signalosome-like subunit and its role in DNA repair. Mol Cell 12: 1087-1099.
- Clark SL, Rodriguez AM, Snyder RR, Hankins GD, Boehning D (2012) Structure-Function Of The Tumor Suppressor BRCA1. Comput Struct Biotechnol J 1.
- Hou YC, Deng JY (2015) Role of E3 ubiquitin ligases in gastric cancer. World J Gastroenterol 21: 786-793.
- Kashima L, Idogawa M, Mita H, Shitashige M, Yamada T, et al. (2012) CHFR protein regulates mitotic checkpoint by targeting PARP-1 protein for ubiquitination and degradation. J Biol Chem 287: 12975-12984.
- Cheung HW, Ching YP, Nicholls JM, Ling MT, Wong YC, et al. (2005) Epigenetic inactivation of CHFR in nasopharyngeal carcinoma through promoter methylation. Mol Carcinog 43: 237-245.
- Laszlo GS, Cooper JA (2009) Restriction of Src activity by Cullin-5. Curr Biol 19: 157-162.
- Wang Z, Inuzuka H, Fukushima H, Wan L, Gao D, et al. (2011) Emerging roles of the FBW7 tumour suppressor in stem cell differentiation. EMBO Rep 13: 36-43.
- Onoyama I, Tsunematsu R, Matsumoto A, Kimura T, de Alborán IM, et al. (2007) Conditional inactivation of Fbxw7 impairs cell-cycle exit during T cell differentiation and results in lymphomatogenesis. J Exp Med 204: 2875-2888.
- Wang Z, Liu P, Inuzuka H, Wei W (2014) Roles of F-box proteins in cancer. Nat Rev Cancer 14: 233-247.
- Cesari R, Martin ES, Calin GA, Pentimalli F, Bichi R, et al. (2003) Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25-q27. Proc Natl Acad Sci U S A 100: 5956-5961.
- Tay SP, Yeo CW, Chai C, Chua PJ, Tan HM, et al. (2010) Parkin enhances the expression of cyclin-dependent kinase 6 and negatively regulates the proliferation of breast cancer cells. J Biol Chem 285: 29231-29238.
- Dawson TM, Dawson VL (2010) The role of parkin in familial and sporadic Parkinson’s disease. Mov Disord 25 Suppl 1: S32-39.
- Lininger RA, Park WS, Man YG, Pham T, MacGrogan G, et al. (1998) LOH at 16p13 is a novel chromosomal alteration detected in benign and malignant microdissected papillary neoplasms of the breast. Hum Pathol 29: 1113-1118.
- Inoue Y, Imamura T (2008) Regulation of TGF-beta family signaling by E3 ubiquitin ligases. Cancer Sci 99: 2107-2112.
- Wang Y, Ren F, Wang Y, Feng Y, Wang D, et al. (2014) CHIP/Stub1 functions as a tumor suppressor and represses NF-κB-mediated signaling in colorectal cancer. Carcinogenesis 35: 983-991.
- Tsai YC, Mendoza A, Mariano JM, Zhou M, Kostova Z, et al. (2007) The ubiquitin ligase gp78 promotes sarcoma metastasis by targeting KAI1 for degradation. Nat Med 13: 1504-1509.
- Goka ET, Lippman ME (2015) Loss of the E3 ubiquitin ligase HACE1 results in enhanced Rac1 signaling contributing to breast cancer progression. Oncogene 34: 5395-5405.
- Yu P, Chen Y, Tagle DA, Cai T (2002) PJA1, encoding a RING-H2 finger ubiquitin ligase, is a novel human X chromosome gene abundantly expressed in brain. Genomics 79: 869-874.
- Paolino M, Choidas A, Wallner S, Pranjic B, Uribesalgo I1, et al. (2014) The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 507: 508-512.
- Ramamoorthy S, Nawaz Z (2008) E6-associated protein (E6-AP) is a dual function coactivator of steroid hormone receptors. Nucl Recept Signal 6: e006.
- [No authors listed] (2013) The MULE/HUWE1 E3 ubiquitin ligase is a tumor suppressor. Cancer Discov 3: OF32.
- Lu G, Zhang Q, Huang Y, Song J, Tomaino R, et al. (2014) Phosphorylation of ETS1 by Src family kinases prevents its recognition by the COP1 tumor suppressor. Cancer Cell 26: 222-234.
- Bromberg KD, Kluger HM, Delaunay A, Abbas S, DiVito KA, et al. (2007) Increased expression of the E3 ubiquitin ligase RNF5 is associated with decreased survival in breast cancer. Cancer Res 67: 8172-8179.
- Cheng H, Wang A, Meng J, Zhang Y, Zhu D (2015) Enhanced metastasis in RNF13 knockout mice is mediated by a reduction in GM-CSF levels. Protein Cell 6: 746-756.
- Ju UI, Park JW, Park HS, Kim SJ, Chun YS (2015) FBXO11 represses cellular response to hypoxia by destabilizing hypoxia-inducible factor-1α mRNA. Biochem Biophys Res Commun 464: 1008-1015.
- Li C, Ao J, Fu J, Lee DF, Xu J, et al. (2011) Tumor-suppressor role for the SPOP ubiquitin ligase in signal-dependent proteolysis of the oncogenic co-activator SRC-3/AIB1. Oncogene 30: 4350-4364.
- Zhang WL, Zhang JH1, Wu XZ2, Yan T3, Lv W3 (2015) miR-15b promotes epithelial-mesenchymal transition by inhibiting SMURF2 in pancreatic cancer. Int J Oncol 47: 1043-1053.
- Zhu J, Zhao C, Kharman-Biz A, Zhuang T, Jonsson P, et al. (2014) The atypical ubiquitin ligase RNF31 stabilizes estrogen receptor α and modulates estrogen-stimulated breast cancer cell proliferation. Oncogene 33: 4340-4351.
- Shinada K, Tsukiyama T, Sho T, Okumura F, Asaka M, et al. (2011) RNF43 interacts with NEDL1 and regulates p53-mediated transcription. Biochem Biophys Res Commun 404: 143-147.
Citation: Jha SK, Jha NK, Kumar P, Ambasta RK (2016) Molecular Chaperones and Ubiquitin Proteasome System in Tumor Biogenesis: An Overview. J Cell Biol Cell Metab 3: 010.
Copyright: © 2016 Saurabh Kumar Jha, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.