Molecular Diagnosis and Prenatal Diagnosis in Abelson’s Helper Integration 1 gene (AHI1) in a Family with Joubert Syndrome
*Corresponding Author(s):
Chen BiliangDepartment Of Obstetrics And Gynecology, Xijing Hospital,Fourth Military Medical University, No. 127, Changle Road (West), Xin Cheng District, Xi’an City, Shaanxi Province, China
Tel:86-29-84771236,
Fax:86-29-84771236
Email:xyfck1010@aliyun.com
Abstract
Keywords
INTRODUCTION
Joubert syndrome (JS, MIM [213300]) was first described in a family in 1968 by Dr. Joubert and his colleagues [1]. The Molar Tooth Sign (MTS) is the key characteristic of JS, it shows unique complex midbrain-hindbrain malformation, appeared in nearly all cases [2]; other diagnostic features include hypotonia in infancy, cerebellar ataxia, intellectual disability and developmental delay, abnormal eye movements and/or breathing pattern [3,4]. According to some literature, autistic has been a comparatively common component of JS [5]. Joubert Syndrome and Related Disorders (JSRD) encompass classic JS and additional features. JSRD are divided into six types [6]: (1) classic JS; (2) JS with ocular defect (JS-O); (3) JS with renal defect (JS-R); (4) JS with oculorenal defects (JS-OR); (5) JS with hepatic defect (JS-H, or COACH syndrome); (6) JS with orofaciodigital defects (JS-OFD, or OFDVI syndrome). The prevalence of JS and JSRD has been estimated at approximately 1:100,000 [7].
According to data, JSRD can be inherited in an autosomal recessive manner and an X-linked manner, and some genes associates with JSRD which are common in clinic are listed in the table 1 (Table 1, Gene Reviews, NCBI). Based on these genes, less than half of the mutations with JSRD can be identified. As a result, molecular diagnosis of these patients and their family are very important.
|
JBTS |
Gene/Protein |
Variation ratio |
Locus |
Numbers of exons |
|
JBTS-1 |
INPP5E |
unknown |
9q34.3 |
10 |
|
JBTS-3 |
AHI1 |
7%-10% |
6q23.2 |
29 |
|
JBTS-4 |
NPHP1 |
1%-2% |
2q13 |
20 |
|
JBTS-5 |
CEP290 |
10% |
12q21.33 |
54 |
|
JBTS-6 |
TMEM67 |
10% |
8q22.1 |
32 |
|
JBTS-7 |
RPGRIP1L |
2%-4% |
16q12.2 |
27 |
|
JBTS-8 |
ARL13B |
<1% |
3q11.1 |
12 |
|
JBTS-9 |
CC2D2A |
10% |
4q15.33 |
38 |
|
JBTS-10 |
OFD1 |
rare |
Xp22 |
26 |
|
JBTS-2 |
TMEM216 |
3% |
11q13.1 |
6 |
|
JBTS-15 |
CEP41 |
<1% |
7q32 |
12 |
|
JBTS-14 |
TMEM237 |
<1% |
2q33 |
13 |
|
JBTS-17 |
C5Orf42 |
unknown |
5p13 |
55 |
|
JBTS-12 |
KIF7 |
unknown |
15q26.1 |
21 |
|
JBTS-13 |
TCTN1 |
unknown |
12q24.11 |
19 |
|
Unassigned |
TCTN2 |
unknown |
12q24.31 |
18 |
|
JBTS-18 |
TCTN3 |
unknown |
10q24.1 |
14 |
|
JBTS-16 |
TMEM138 |
unknown |
11q12.2 |
6 |
|
JBTS-20 |
TMEM231 |
unknown |
16q23.1 |
7 |
|
JBTS-11 |
TTC21B |
unknown |
2q24.3 |
30 |
Table 1: Genes and loci causative of JSRD.
In this article, we discussed a Chinese patient of JSRD and the prenatal diagnosis for the proband’s family. This proband has been actually diagnosed JSRD. We used Sanger to identify variants in this proband and found compound heterozygosity for one novel mutation in exon 18 of gene AHI1:c.2386_c.2380delTTTC and one known mutation in exon 8 of gene AHI1:c.910_911insA.
METHODS
Case report
In the department of Obstetrics and Gynecology of the first affiliated hospital of The Fourth Military Medical University, we received a Chinese family with JS proband. The proband’s mother was pregnant for 18 weeks (Figure 1a), when she visited our hospital. The proband with clinical manifestations of Joubert syndrome got supportive brain imaging in the Department of Radiology, Maternal and Child Health Hospital, in Shanxi province.
The proband, a three months old male Chinese baby, who was born at term naturally without any complications, was the first child of his healthy parents. His mother denied consanguinity with her husband and had no history of teratogen exposure during or immediately preceding pregnancy.
Clinical examination was undertaken when the boy is 3 months old and the boy was died when he is six months old. Hypotonia and abnormal right eye movement is observed. Developmental delayed was also noticed, meantime. Magnetic Resonance Imaging (MRI) of the brain showed Cerebellar Vermis Hypoplasia (CVH), deepened interpeduncular fossa, thick and elongated superior cerebellar peduncles, abnormally shaped fourth ventricle and hypoplastic vermis. (Figure 1b). All the features showed classical MTS, which are the key diagnostics criteria of JSRD clinically.
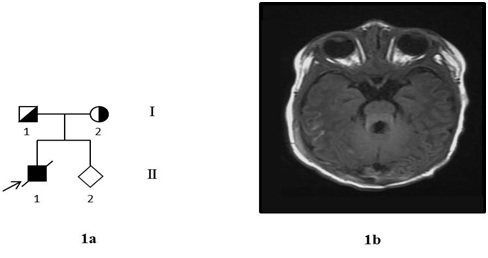 Figure 1: 1a. Pedigree drawing of the proband’s family.
Figure 1: 1a. Pedigree drawing of the proband’s family.1b. MRI image of the brain of the proband with Joubert syndrome showing a typical Molar Tooth Sign (MTS).
Targeted next-generation sequencing and variants verify
RESULTS
In this study, the exons of 20 genes related to Joubert syndrome and related disorders were recognized by next generation sequencing and detected by DNA sequencing. Maternal effect has been eliminated by Short Tandem Repeat (STR). Approximately 158 kb of the “target region” for the proband were captured and sequenced. The average coverage is 99.58% for the family, and the average sequencing depth is 463.25-fold.
Only two potential mutations in AHI1 were detected from the proband: c.910_911insA in the exon 8 and c.2323_2326delGAAA in the exon 18, and both of them are in the coding region. Because no more DNA of the proband can verify the result offered by targeted next-generation sequencing, Sanger sequencing technology was employed for the parents in order to identify the potential mutations and reveal the inheritance background of JSRD for this family (Figure 2). According to the analysis, the truncating mutation (c.910_911insA) was inherited from the proband’s mother, which has been reported [9]. The frame shift (c.2323_2326delGAAA) was found both in the proband and his father, which is novel. The result shows that the proband’s parents are both carriers.
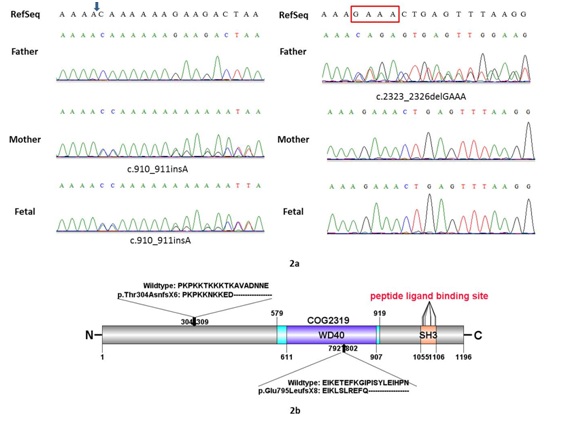 Figure 2: a: Sanger sequencing verification of the mutations identified by next-targeting sequence.
Figure 2: a: Sanger sequencing verification of the mutations identified by next-targeting sequence.
b: The predicated full-length human Joubert in protein gives a 1196-aa message with WD repeats and SH3 domain. The arrows indicate the relative position of the mutations detected.
The DNA extracted from amniotic fluid cells were amplified by PCR and detected by DNA sequencing. The data shows all the JS and JSRD-related genes have been amplified and analyzed and the fetus inherits thec.910_911insA in the exon 8 from her mother. DNA extracted from cord blood validates our testing result.
DISCUSSION
In our study, we identified the genetic base of this Chinese male baby with Joubert syndrome, and found compound heterozygosity for one novel mutation in gene AHI1, and carry out the prenatal diagnosis for the proband’s family. The genes caused JS and JSRD uncover two major phenomenon: (1) all the genes coding for proteins of the primary cilium were known as “ciliopathies”; (2) some genes were concerned with the clinical phenotypes. So far, AHI1 is the most commonly mutated gene and has only found in the JSRD [10-13], nearly 10-15% of all patients have mutations in AHI1, and associated with autism [5]. The most frequently feature in AHI1-realated JSRD is retinal dystrophy existing in almost 80% patients with mutations in AHI1. According to the data, this proband should be JSRD instead of classical JS.
AHI1 mRNA is expressed highly in brain and kidney in human fetal tissue, and in cerebellum and cerebral cortex in human adult, respectively. To the contrary, it is expressed weakly in liver and lung in human fetal tissue and in the putamen in human adult, separately [14]. AHI1, which encodes cytoplasmic adaptor protein, is composed of an N-terminal coiled-coil domain, seven WD40 repeats and one SH3 domain [15].
The truncating mutation, c.910_911insA (p.Thr304AsnfsX6) in exon 8, which was first described by a Duth research group, causes frame shift and damaging effect on AHI1 protein function. Retinal disease, which also performed in this Chinese boy, showed in nearly 76% in AHI1-related JSRD patient [9, 11-14]. Because of the deletion identified in exon 18 of the gene AHI1 causes frame shift and premature stop codon, it was predicted to be destructive. These four-base deletion (c.2323_2326delGAAA) in the exon18 truncating the protein at amino acid 802 (p.Glu795LeufsX8) and also eliminating the WD repeats and SH3 domain.
Because of the fetus is a carrier of JSRD, genetic counseling should be carried out when the baby grow up, get married and be ready to have a baby. As molecular diagnosis is the precise criterion of JS and JSRD, we suggest all JS and JSRD-related genes mutation analysis should be performed in prenatal diagnosis, even if the pathogenic mutation of the proband have been identify. This is an effective mean to avoid the result of false-positive or false-negative.
In this case, the combination of target gene capture, next-generation, polymerase chain reaction and Sanger sequencing were applied in order to verify the gene mutations in the Chinese male baby with Joubert syndrome and conduct the prenatal diagnosis for his family. Our finding gives a description of the variation in gene AHI1, includes one novel mutation, and indicted the importance of molecular diagnosis and prenatal diagnosis for the families with recessively inherited disorders.
REFERENCES
- Joubert M, Eisenring JJ, Andermann F (1968) Familial dysgenesis of the vermis: a syndrome of hyperventilation, abnormal eye movements and retardation. Neurology 18: 302-303.
- Maria BL, Hoang KB, Tusa RJ, Mancuso AA, Hamed LM, et al. (1997) "Joubert syndrome" revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol 12: 423-430.
- van Dorp DB, Palan A, Kwee ML, Barth PG, van der Harten JJ (1991) Joubert syndrome: a clinical and pathological description of an affected male and a female fetus from the same sibship Am J Med Genet 40: 100-104.
- Chance PF, Cavalier L, Satran D, Pellegrino JE, Koenig M, et al. (1999) Clinical nosologic and genetic aspects of Joubert and related syndromes. J Child Neurol 14: 660-672.
- Alvarez Retuerto AI, Cantor RM, Gleeson JG, Ustaszewska A, Schackwitz WS, et al. (2008) Association of common variants in the Joubert syndrome gene (AHI1) with autism. Hum Mol Genet 17: 3887-3896.
- Valente EM, Dallapiccola B, Bertini E (2013) Joubert syndrome and related disorders. Handb Clin Neurol 113: 1879-1888.
- Yachnis AT, Rorke LB (1999) Neuropathology of Joubert syndrome. J Child Neurol 14: 655-659.
- Wei X, Ju X, Yi X, Zhu Q, Qu N, et al. (2011) Identification of sequence variants in genetic disease-causing genes using targeted next-generation sequencing. PLoS One; 6: 29500.
- Kroes HY, van Zon PH, Fransen van de Putte D, Nelen MR, Nievelstein RJ, et al. (2008) DNA analysis of AHI1, NPHP1 and CYCLIN D1 in Joubert syndrome patients from the Netherlands. Eur J Med Genet 51: 24-34.
- Parisi MA (2009) Clinical and molecular features of Joubert syndrome and related disorders. Am J Med Genet C Semin Med Genet 151: 326-340.
- Dixon-Salazar T, Silhavy JL, Marsh SE, Louie CM, Scott LC, et al. (2004) Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet 75: 979-987.
- Utsch B, Sayer JA, Attanasio M, Pereira RR, Eccles M, et al. (2006) Identification of the first AHI1 gene mutations in nephronophthisis-associated Joubert syndrome. Pediatr Nephrol 21: 32-35.
- Valente EM, Brancati F, Silhavy JL, Castori M, Marsh SE, et al. (2006) AHI1 gene mutations cause specific forms of Joubert syndrome-related disorders. Ann Neurol 59: 527-534.
- Ferland RJ, Eyaid W, Collura RV, Tully LD, Hill RS, et al. (2004) Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet 36: 1008-1013.
- Jiang X, Hanna Z, Kaouass M, Girard L, Jolicoeur P (2002) Ahi-1, a novel gene encoding a modular protein with WD40-repeat and SH3 domains, is targeted by the Ahi-1 and Mis-2 provirus integrations. J Virol 76: 9046-9059.
Citation: Biliang C, Yu L, Ying X, Jianfang Z, Xing T, et.al. (2016) Molecular Diagnosis and Prenatal Diagnosis in Abelson’s Helper Integration 1gene (AHI1) in a Family with Joubert Syndrome. J Reprod Med Gynecol Obstet 1: 003.
Copyright: © 2016 Chen Biliang, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.