Multiple Fetal Anomalies: A Case of Complete Triploidy
*Corresponding Author(s):
Holman JLNDepartment Of Neonatology, Brenner Children’s Hospital, Wake Forest Baptist Medical Center, Winston-Salem, North Carolina, United States
Tel:+1 3367130629,
Email:jholman@wakehealth.edu
Abstract
Complete triploidy is a rare genetic condition characterized by an additional complete chromosome set within all cells. Its presentation is typically lethal, although case reports of infants with partial or complete triploidy surviving hours to days exist. Presentation is associated with multiple congenital anomalies. We herein report a rare presentation of triploidy in a neonatal patient who suffered from prematurity, respiratory distress, metabolic acidosis and subsequently expired. Diagnostic tests ultimately revealed complete triploidy. In spite of what is understood about the genetics of the condition, options for the management of this disorder are not well described in the literature, as cases are extremely rare.
CASE REPORT
A 0.9 kg infant is born to a 31-year-old G3P2 female via urgent c-section at 28.6 weeks gestation due to worsening pre-eclampsia. Pregnancy complicated by oligohydramnios, thickened globular placenta which obscures anatomical survey on both ultrasound and fetal MRI (Figures 1-3), abnormal non-invasive prenatal screening and multiple congenital anomalies including: IUGR, hypoplasia of fetal nasal bone, prominent enlarged liver, pericardial effusion with VSD, and asymmetric fetal cerebral ventricles. Mother declines amniocentesis. Mother’s serologies are negative. At delivery, infant has poor respiratory effort and requires intubation. Apgar scores are 3, 7 and 8 at 1, 5 and 10 minutes. Infant is admitted to the Neonatal Intensive Care Unit (NICU) on conventional ventilator.
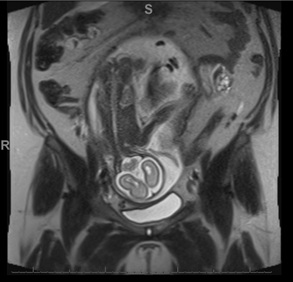
Figure 1: The placenta is enlarged and heterogeneous. The appearance is nonspecific and may be secondary to report triploidy and/or partially due to maternal hypertension. Fetal assessment is limited secondary to the enlarged placenta causing mass effect displacing the fetus towards the right as well as limited due to oligohydramnios.
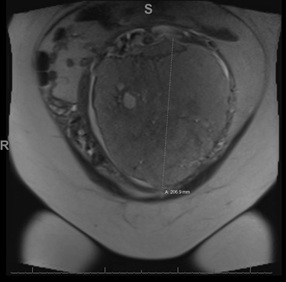
Figure 2: The more focal T2 hypointense region about the left side of the thickened placenta is nonspecific, though may be sequelae of prior placental hemorrhage. The placenta is enlarged and heterogeneous. The appearance is nonspecific and may be secondary to report triploidy and/or partially due to maternal hypertension. Some heterogeneity of the placenta is expected later in pregnancy, though this degree of heterogeneity is more than expected at the gestational age.
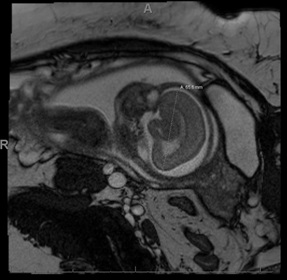
Figure 3: Fetal assessment is limited secondary to the enlarged placenta causing mass effect displacing the fetus towards the right as well as limited due to oligohydramnios. Within the limitations of the study, the fetus measures small for age, with calvarial/intracranial measurements all lower than expected for gestational age. The cavum septum pellucidum is not clearly visualized, and the gyral/sulcation pattern appears less developed than expected for gestational age. There also is slight prominence of the extra-axial spaces throughout the exam.
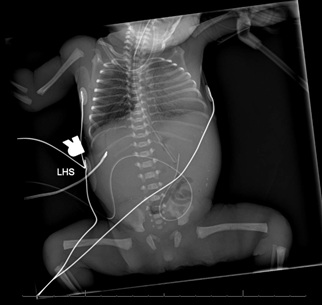
Figure 4: Diffuse interstitial opacification likely reflecting pulmonary edema vs. RDS. Scattered calcifications in the abdomen concerning for meconium peritonitis.
Physical examination reveals an infant who is intubated, has widely separated sutures, small mouth, low set ears, ambiguous genitalia, brachydactylic hands, and single transverse palmar crease on the right.
Chest x-ray shows expansion of 8 ribs and appropriate placement of endotracheal tube (Figure 4). Capillary blood gas reveals pH 7.11, pCO2 86, pAO2 48, HCo3 27 and base deficit -3.Na 140. Karyotype and microarray are sent.
PATIENT COURSE
Infant is given one dose of surfactant and an umbilical venous catheter is placed. Infant is treated for hypoglycemia. SpO2 levels and heart rate continue to decline despite treatment with escalation to High Frequency Ventilation (HFJV), PPV, fluid resuscitation and epinephrine administration. Infant continues to deteriorate despite maximal resuscitative efforts as CPR is initiated. Parents decide, given patient’s lack of response to resuscitation and multiple congenital anomalies, to transition to comfort care. Infant dies in family’s arms at approximately 9 hours of life.
DISCUSSION AND CONCLUSION
Differential diagnosis: Partial/mosaic or complete triploidy, trisomy 13/18, other aneuploidy, severe IUGR due to placental insufficiency.
Actual diagnosis: Karyotype returns with 69XXY, consistent with triploidy.
CONDITION
Triploidy is a sporadic, rare, typically lethal chromosomal anomaly characterized by an additional haploid chromosome set. It occurs in 2-3% of recognized human conceptions and accounts for roughly 20% of spontaneous abortions related to chromosomal abnormalities in the first trimester [1,2]. The extra set of chromosomes in triploidy is a cause of a variety of serious birth defects, placental problems, molar anomalies, and severe growth problems in a fetus, although there can be marked variability in the presence of these anomalies [1,3]. Infants with this lethal condition are generally small due to severe Intrauterine Growth Retardation (IUGR) and they have multiple congenital defects, including facial abnormalities, micrognathia, cleft lip, heart defects, neural tube defects (spina bifida), limb abnormalities and other serious congenital anomalies of the kidney and umbilical cord, including single umbilical artery [1,4]. Live birth of true triploid infants is rare, although case reports exist-survival is generally limited in these cases to hours or days [1,3].
Parental origin of the extra genome set in triploidy has specific effects on both fetal phenotype and placental development due to imprinting effect [5]. An extra haploid chromosome set can be paternal (diandric) or maternal (digynic) in origin. There are different mechanisms that produce triploidy: Spontaneous fertilization of ovum by 2 sperm, normal fertilization of ovum by diploid sperm (diandry), or fertilization of primary oocyte, or diploid oocyte that is product of nondisjunction during meiosis or retention of polar body (digyny) [1,5].
Diagnosis can be made prenatally by cytogenetic analysis of cells obtained through amniocentesis [1]. The diagnosis of triploidy can also be supported by elevated fetal nuchal translucency, as well as elevated maternal serum Alpha Fetoprotein (AFP) and total and beta-human Chorionic Gonadotropin (hCG) levels and low maternal serum Pregnancy-Assisted Plasma Protein-A (PAPP-A) levels in the first trimester [1,4,6]. Fetal ultrasound during the first and second trimesters is important for identification of congenital anomalies and growth restriction [1]. After birth, diagnosis can be confirmed by blood chromosome analysis (microarray, karyotype), or Fluorescent in Situ Hybridization (FISH). There is no treatment or cure for triploidy [6].
LESSONS FOR THE CLINICIAN
• Although there is no constellation if symptoms that is pathognomonic for triploidy, the condition should be suspected in cases of major congenital malformations that involve multiple systems and significant growth restriction that presents early in gestation and persists.
• Microarray and karyotyping offer the opportunity to identify chromosomal abnormalities in the presence of other congenital anomalies.
FUNDING
This case report received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
DECLARATION OF CONFLICTING INTEREST
The Authors declare that there are no conflicts of interest.
REFERENCES
- Kolarski M, Ahmetovic B, Beres M, Topic R, Nikic V, et al. (2017) Genetic counseling and prenatal diagnosis of triploidy during the second trimester of pregnancy. Med Arch 71: 144-147.
- Wick JB, Johnson KJ, O'Brien J, Wick MJ (2013) Second-trimester diagnosis of triploidy: A series of four cases. AJP Rep 3: 37-40.
- Fraikor AL, Vigneswaran R, Honeyfield P (1980) Live-born triploid. Am J Dis Child 134: 988-989.
- Toufaily MH, Roberts DJ, Westgate MN, Holmes LB (2016) Triploidy: Variation of phenotype. Am J Clin Pathol 145: 86-95.
- McFadden DE, Kalousek DK (1991) Two different phenotypes of fetuses with chromosomal triploidy: correlation with parental origin of the extra haploid set. Am J Med Genet 38: 535-538.
- Fergus KA, et al. (2016) Triploidy. In: Moy T, Avery L (eds.). The gale encyclopedia of genetic disorders, (4th edn). Gale, Michigan, United States. Pg no: 1751-1755.
Citation: Holman JLN, McGowan MEB (2020) Multiple Fetal Anomalies: A Case of Complete Triploidy. J Neonatol Clin Pediatr 7: 045.
Copyright: © 2020 Holman JLN, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.